Practice Essentials
Polycythemia vera (PV) is a stem cell disorder characterized as a panhyperplastic, malignant, and neoplastic marrow disorder. Its most prominent feature is an elevated absolute red blood cell mass because of uncontrolled red blood cell production. This is accompanied by increased white blood cell (myeloid) and platelet (megakaryocytic) production, which is due to an abnormal clone of the hematopoietic stem cells with increased sensitivity to the different growth factors for maturation. [1, 2, 3, 4]
Signs and symptoms of polycythemia vera
Impaired oxygen delivery due to sludging of blood may lead to the following symptoms:
-
Headache
-
Dizziness
-
Vertigo
-
Tinnitus
-
Visual disturbances
-
Angina pectoris
-
Intermittent claudication
Bleeding complications, seen in approximately 1% of patients with PV, include epistaxis, gum bleeding, ecchymoses, and gastrointestinal bleeding. Thrombotic complications (1%) include venous thrombosis or thromboembolism and an increased rate of stroke and other arterial thromboses.
Physical examination findings may include the following:
-
Splenomegaly (75% of patients)
-
Hepatomegaly (30%)
-
Plethora
-
Hypertension
Diagnosis of polycythemia vera
According to 2022 revised World Health Organization (WHO) guidelines, diagnosis of PV requires requires the presence of either all three major criteria or the first two major criteria and the minor criterion. [5]
Major WHO criteria are as follows:
Hemoglobin > 16.5 g/dL in men and > 16 g/dL in women, or hematocrit > 49% in men and > 48% in women
Bone marrow biopsy showing hypercellularity for age with trilineage growth (panmyelosis) including prominent erythroid, granulocytic, and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size)
Presence of JAK2V617F or JAK2 exon 12 mutation
The minor WHO criterion is as follows:
-
Serum erythropoietin level below the reference range for normal
Previous versions of the WHO guidelines included red cell mass > 25% above mean normal predicted value as a diagnostic criterion. The WHO removed this criterion from its 2022 guidelines because the determination of red cell mass with chromium-51–labeled red cells has become uncommon in routine clinical practice. [5]
Management of polycythemia vera
Treatment measures are as follows:
-
Phlebotomy – To keep hematocrit below 45%
-
Aspirin – 81 mg daily
-
Cytoreductive therapy – For patients at high risk for thrombosis
-
Splenectomy – In patients with painful splenomegaly or repeated episodes of splenic infarction
Hydroxyurea is the most commonly used cytoreductive agent. If hydroxyurea is not effective or not tolerated, alternatives include the following:
-
Ropeginterferon alfa 2b
-
Busulfan – In patients older than 65 years
-
Ruxolitinib (Jakafi)
-
Fedratinib (Inrebic)
For discussion of polycythemia in children, see Pediatric Polycythemia vera.
Pathophysiology
The bone marrow of patients with polycythemia vera (PV) contains normal stem cells but also contains abnormal clonal stem cells that interfere with or suppress normal stem cell growth and maturation. The panmyelosis in PV appears to result from unregulated neoplastic proliferation. The origin of the stem cell transformation remains unknown. See the image below.
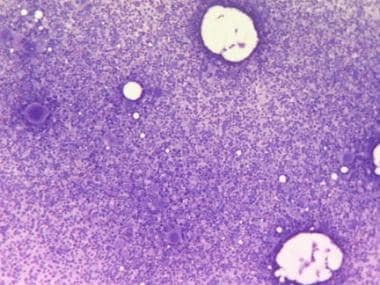 Bone marrow film at 100X magnification demonstrating hypercellularity and increased number of megakaryocytes. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Bone marrow film at 100X magnification demonstrating hypercellularity and increased number of megakaryocytes. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Progenitors of the blood cells in these patients display abnormal responses to growth factors, suggesting the presence of a defect in a signaling pathway common to different growth factors. The observation that in vitro erythroid colonies grow when no endogenous erythropoietin (Epo) is added to the culture and the presence of a truncated Epo receptor in familial erythrocytosis indicate that the defect is in the transmission of the signal. The sensitivity of PV progenitors to multiple cytokines suggests that the defect may lie in a common pathway downstream from multiple receptors. Increased expression of BCLX suggests an additional decrease in cellular apoptosis.
A mutation of the Janus kinase–2 gene (JAK2) is the most likely source of PV pathogenesis, as JAK2 is directly involved in the intracellular signaling following exposure to cytokines to which PV progenitor cells display hypersensitivity. [6] A recurrent unique acquired clonal mutation in JAK2 has been found in most patients with PV and other myeloproliferative diseases (MPDs), including essential thrombocythemia and idiopathic myelofibrosis.
A unique valine-to-phenylalanine substitution at position 617 (V617F) in the pseudokinase JAK2 domain has been identified. The substitution, called JAK2V617F, leads to a permanently turned-on signaling at the affected cytokine receptors. [7, 8, 9, 10] The JAK2V617F mutation is present in more than 95% of PV cases, but is also found in 50-60% of essential thrombocytosis and primary myelofibrosis cases. [11] How these mutations interact with the wild-type kinase genes and how they manifest into different forms of MPDs need to be elucidated.
At diagnosis of PV, a homozygous JAK2 genotype is found less often in women than in men (median, 61% vs 80%). JAK2 variant allele frequency, which is initially similar in men and women, over time becomes significantly higher in men than in women. [12]
Thrombosis and bleeding are frequent in persons with PV, as a result of the disruption of hemostatic mechanisms because of (1) increased numbers of red blood cells and (2) elevation of the platelet count. There are findings that indicate the additional roles of tissue factor and polymorphonuclear leukocytes (PMLs) in clotting, the platelet surface as a contributor to phospholipid-dependent coagulation reactions, and the entity of platelet microparticles. Tissue factor is also synthesized by blood leukocytes, the level of which is increased in persons with MPD, which can contribute to thrombosis.
PV tends to be milder in women than in men, with lower rates of myocardial infarction and peripheral arterial disease (although this may be related to lower rates of smoking in women). However, venous thrombosis is more common in females; in particular, the rate of splanchnic vein thrombosis is significantly higher in young women. [12]
Rusak et al evaluated the hemostatic balance in patients using thromboelastography and also studied the effect of isovolemic erythrocytapheresis on patients with PV. They concluded that thromboelastography may help to assess the thrombotic risk in these patients. [13]
Hyperhomocystinemia is a risk factor for thrombosis and is also widely prevalent in patients with MPD (35% in controls, 56% in persons with PV).
Acquired von Willebrand syndrome is an established cause of bleeding in persons with MPD, accounting for approximately 12-15% of all patients with this syndrome. von Willebrand syndrome is largely related to the absorption of von Willebrand factor onto the platelets; reducing the platelet count should alleviate the bleeding from the syndrome.
Etiology
The causes of polycythemia vera (PV) are unknown, but a number of approaches are being studied to define the molecular lesion or lesions. The JAK2 V617F mutation can give rise to a turned-on cytokine receptor, leading to pancytosis similar to the PV phenotype. This is similar to the biologic properties of the BCR/ABL abnormality in that they both mimic cytokine signaling.
Clonality studies using a rare polymorphism in the G6PD gene demonstrate predominant expression of a single allele in all blood cell lines. X-chromosome inactivation studies have played a pivotal role in establishing current concepts of many hematologic malignancies. Approximately 90% of patients with PV show a skewed pattern of X inactivation in all their blood cell lines, indicating support for the concept of a transformed multipotential stem cell.
Cytogenetic studies show the presence of an abnormal karyotype in the hematopoietic progenitor cells in approximately 34% of patients with PV, depending on the stage of the disease in which the study was performed. Approximately 20% of patients have cytogenetic abnormalities at diagnosis, increasing to more than 80% for those with more than 10 years of follow-up care.
The following genetic abnormalities, which are similar to the abnormal karyotypes observed in patients with myelodysplastic syndromes and other MPDs, have been observed in patients with PV:
-
Deletion of 20q (8.4%)
-
Deletion of 13q (3%)
-
Trisomy 8 (7%)
-
Trisomy 9 (7%)
-
Trisomy of 1q (4%)
-
Deletion of 5q or monosomy 5 (3%)
-
Deletion of 7q or monosomy 7 (1%)
Spivak and colleagues analyzed gene expression in CD34+ peripheral blood cells from 19 patients with PV and found twice as many up-regulated or down-regulated genes in men as in women. In addition, these researchers found 102 genes with differential regulation that was concordant in men and women and that could be used to divide patients into two phenotypical groups. The groups differed significantly with respect to disease duration, clinical manifestations, and prognosis. [14]
Epidemiology
Frequency
United States
Polycythemia vera (PV) is relatively rare, occurring in 0.6-1.6 persons per million population.
Race-, sex-, and age-related demographics
Originally, Ashkenazi Jewish persons were thought to have a higher predilection for polycythemia vera than members of other ethnic groups. Subsequently, however, many studies have shown that this condition occurs in all ethnic groups.
Most studies have found the incidence of polycythemia vera to be slightly higher in males than females. However, a slightly higher incidence in females has also been reported, and one systematic review and meta-analysis showed no significant difference in the crude annual incidence between males and females. [12]
The peak incidence of polycythemia vera onset is age 50-70 years. However, this condition occurs in persons of all age groups, including early adulthood and childhood, albeit rarely.
Prognosis
Median survival in patients with polycythemia vera (PV), which is 1.5-3 years in the absence of therapy, has been extended to approximately 14 years overall, and to 24 years for patients younger than 60 years of age, because of new therapeutic tools. [15] However, according to a study of Surveillance, Epidemiology and End Results (SEER) data, mortality in PV patients is higher than in an age- and sex-matched population. Five-year survival in the overall cohort was 79.5% but patients are at a high risk of second primary malignancies and leukemic transformation, which may compromise long-term survival. [16]
Morbidity and mortality
The major causes of morbidity and mortality are as follows:
-
Thrombosis
-
Hemorrhage
-
Peptic ulcer disease
-
Myelofibrosis
-
Acute leukemia or myelodysplastic syndrome
Thrombosis
Venous and arterial thrombosis has been reported in 15-60% of patients, depending on the control of their disease. It is the major cause of death in 10-40% of patients. All of the following have been noted:
-
Pulmonary embolism
-
Kidney failure from renal vein or artery thrombosis
-
Intestinal ischemia from mesenteric vein thromboses
-
Peripheral arterial emboli
Hemorrhage
Hemorrhagic complications occur in 15-35% of patients and lead to death in 6-30% of these patients. Bleeding is usually the consequence of vascular compromise resulting from ischemic changes from thrombosis or hyperviscosity.
Peptic ulcer disease
Peptic ulcer disease is reported to be associated with PV at a 3- to 5-fold higher rate than that of the general population. This has been attributed to increased histamine serum levels.
Myelofibrosis
Myelofibrosis and pancytopenia occur in 3-10% of patients, usually late in the disease, which is considered the spent phase of PV. In these patients, infections and bleeding complications may be the most serious health threats, and red blood cell transfusions may be required to maintain adequate red blood cell counts and to improve fatigue and other anemia-related symptoms.
The US Food and Drug Administration (FDA) has approved two Janus kinase (JAK) inhibitors for treatment of post-PV myelofibrosis. The JAK1 and JAK2 inhibitor ruxolitinib (Jakafi) was approved in 2011; the highly selective JAK2 inhibitor fedratinib (Inrebic) was approved in 2019. [17]
Leukemia and myelodysplastic syndrome
Acute leukemia or a myelodysplastic syndrome develops in 1.5% of patients treated with phlebotomy alone. The transformation risks increase to 13.5% within 5 years with treatment using chlorambucil and to 10.2% within 6-10 years in patients treated with phosphorus-32. At 15 years, the transformation risk for patients treated with hydroxyurea is 5.9%, which, although not statistically significant, is a worrisome trend.
Abdulkarim et al studied the long-term (15 years) rate of transformation to acute myelogenous leukemia (AML) in Swedish and French patients with Philadelphia chromosome–negative MPD, including 317 patients with PV. The annual rate of AML transformation was 0.38% in patients with PV, and the average time from PV diagnosis to AML transformation was 88 +/- 56 months. Notably, 17 of the 18 patients with PV whose condition transformed to AML were females, despite the fact that almost half of the patients with PV were males. [18]
-
Bone marrow film at 100X magnification demonstrating hypercellularity and increased number of megakaryocytes. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Blood film at 400X magnification demonstrating polyglobulia and thrombocytosis. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Bone marrow film at 400X magnification demonstrating dominance of erythropoiesis. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
This blood film at 10,000X magnification shows a giant platelet and an eosinophil. Erythrocytes show signs of hypochromia as a result of repeated phlebotomies. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Tables

What would you like to print?
- Rusfertide: First Self-Injected Drug to Cut Phlebotomy
- High in the Andes, a Possible Clue Into Blood Cancer Emerges
- Colorectal Cancer Linked With Adverse Sexual Health Outcomes
-
 Can We Prevent Myelofibrosis? The New Interferon Era
Can We Prevent Myelofibrosis? The New Interferon Era
-
JAK Inhibitors: Too Many Choices or Not Enough?
-
Diet, Exercise, and Myelofibrosis: A Holistic Approach



