Background
Optic atrophy is the final common morphologic endpoint of disease process that causes degeneration of axons of the ganglion cells. [1] Clinically, optic atrophy manifests as changes in the color and the structure of the optic disc (cupping) associated with variable degrees of visual dysfunction. The term "atrophy" is a misnomer, as, in its strict histologic definition, atrophy implies involution of a structure due to prolonged disuse. [1]
Optic atrophy may result from damage occurring inside the eye (such as glaucoma, optic neuritis, or papilledema), along the optic nerve pathway to the brain (due to a tumor, neurodegenerative disorder, or trauma), or it may be present from birth (as seen in conditions like Leber's hereditary optic atrophy or autosomal dominant optic atrophy).
Children
Optic atrophy remains a prominent cause of visual impairment in the pediatric population, with its etiologic landscape shifting notably since the turn of the 21st century. [2] Enhanced diagnostic capabilities through advanced neuroimaging and optical coherence tomography have deepened our comprehension of this condition. A notable trend in the new millennium is the rising prevalence of optic atrophy associated with prematurity, correlating with increased rates of premature births and the enhanced survival of these infants due to advancements in neonatal care.
Pathophysiology
The optic nerve comprises approximately 1.2 million axons that originate at the ganglion cell layer of the retina. Upon exiting the eyeball, the axons are covered with myelin sheath provided by oligodendrocytes. Once injured they do not regenerate. Thus, the optic nerve behaves more like a white matter tract rather than a true peripheral nerve.
The optic nerve is divided into the following four parts:
-
Intraocular part (1 mm) (optic nerve head)
-
Intraorbital part (25-30 mm)
-
Intracanalicular part (5-10mm)
-
Intracranial part (10-16 mm)
The average optic nerve head is 1 mm deep, 1.5 mm wide, 1.8 mm deep at the retinal level. The optic nerve head sits at a major transition between an area of high pressure to an area of low pressure (intracranial pressure) and is composed of four types of cells: ganglion cell axons, astrocytes, capillary-associated cells, and fibroblasts.
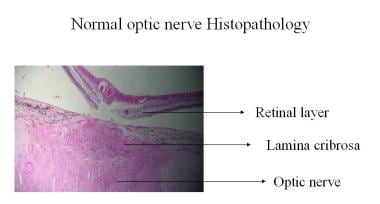
Clinically, the light incident from the ophthalmoscope undergoes total internal reflection through the axonal fibers, and subsequent reflection from the capillaries on the disc surface gives rise to the characteristic yellow-pink color of a healthy optic disc. Degenerated axons lose this optical property, explaining the pallor in optic atrophy.
The blood supply at the optic nerve head is provided by pial capillaries arising from the circle of Zinn-Haller. Alternatively, the loss of these capillaries can also be a cause to pale-appearing disc. The Kestenbaum capillary number index is the number of capillaries counted on the optic disc, which is normally around 10. Less than 6 indicates atrophy and more than 12 indicates hyperemic disc.
Histopathologic changes noted in optic atrophy include the following:
-
Shrinkage or loss of both myelin and axis cylinders
-
Glial cell proliferation
-
Deepening of the physiologic cup with barring of the lamina cribrosa
-
Widening of the subarachnoid space with redundant dura with widening of the pial septa
-
Severed nerve leads to bulbous axonal swellings (Cajal end bulbs); may be observed at the anterior cut end of the fibers
Classification
Optic atrophy is classified as pathologic, ophthalmoscopic, or etiologic.
1. Pathologic optic atrophy
Anterograde degeneration (Wallerian degeneration) - Degeneration begins in the retina and proceeds toward the lateral geniculate body (eg, toxic retinopathy, chronic simple glaucoma). Larger axons disintegrate more rapidly than smaller axons.
Retrograde degeneration - Degeneration starts from the proximal portion of the axon and proceeds toward the optic disc (eg, optic nerve compression by intracranial tumor).
Trans-synaptic degeneration - In trans-synaptic degeneration, a neuron on one side of a synapse degenerates as a consequence of the loss of a neuron on the other side (eg, in individuals with occipital damage incurred either in utero or during early infancy).
2. Ophthalmoscopic optic atrophy
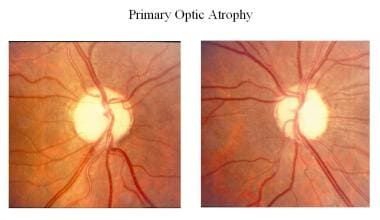
a) Primary optic atrophy
In conditions with primary optic atrophy (eg, pituitary tumor, optic nerve tumor, traumatic optic neuropathy, multiple sclerosis), optic nerve fibers degenerate in an orderly manner and are replaced by columns of glial cells without alteration in the architecture of the optic nerve head. The disc is chalky white with sharply demarcated margins, and the retinal vessels are normal. The lamina cribrosa is well defined.
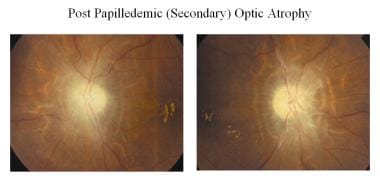
b) Secondary optic atrophy
In conditions with secondary optic atrophy (eg, papilledema, papillitis), the atrophy is a result of disc edema (shown in the image below). Optic nerve fibers exhibit marked degeneration, with excessive proliferation of glial tissue. The surface architecture is lost, resulting in indistinct margins. The disc appears grey or dirty grey, with poorly defined margins. The lamina cribrosa is obscured due to proliferating fibroglial tissue. Hyaline bodies (corpora amylacea) or drusen may sometimes be observed.
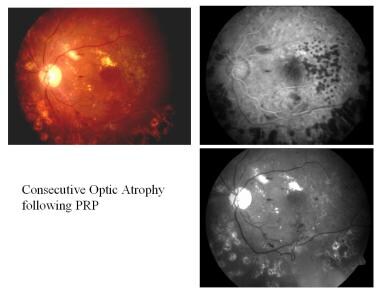
c) Consecutive optic atrophy
Consecutive atrophy is an ascending type of atrophy (eg, chorioretinitis, pigmentary retinal dystrophy, cerebromacular degeneration) that usually results from diseases of the choroid or the retina. The disc is waxy pale with normal disc margins and marked attenuation of arteries.
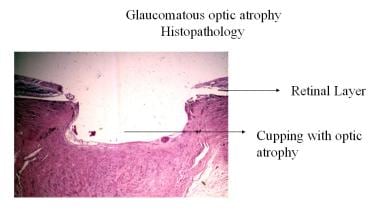
d) Glaucomatous optic atrophy
Also known as cavernous optic atrophy, there is marked cupping of the disc. The main features include vertical enlargement of the optic cup, visibility of the laminar pores (laminar dot sign) with backward bowing of the lamina cribrosa, bayoneting or nasal shifting of the retinal vessels, and peripapillary halo and atrophy.
e) Temporal pallor
Temporal pallor may be observed in traumatic or nutritional optic neuropathy, and it is most commonly seen in patients with multiple sclerosis, particularly in those with a history of optic neuritis. The disc is pale with a clear, demarcated margin and normal vessels, and the physiologic pallor temporally is more distinctly pale.
3. Etiologic optic atrophy
Hereditary atrophy
This is divided into congenital or infantile optic atrophy (recessive or dominant form), Behr hereditary optic atrophy (autosomal recessive), and Leber optic atrophy. [7, 8] Several hereditary optic neuropathies, including optic atrophy type 1 and Leber optic atrophy, have been attributed to mitochondrial dysfunction in RGCs. [1]
Autosomal-dominant optic atrophy type 1 is caused by mutations in the OPA1 gene on chromosome 3q29. The OPA1 protein produced plays a key role in a process called oxidative phosphorylation and in self-destruction of cells (apoptosis). OPA1 is an integral pro-fusion protein within the internal mitochondrial membrane. Mutations in the OPA1 gene lead to vision problems experienced by people with breakdown of structures that transmit visual information from the eyes to the brain. Affected individuals first experience a progressive loss of nerve cells within the retina, RGCs. The loss of these cells is followed by the degeneration (atrophy) of the optic nerve.
X-linked optic atrophy type 2 is caused by mutation in the OPA2 gene with cytogenetic location Xp11.4-p11.21. The patient presents with early-onset childhood vision loss with slow progression of loss.
Hereditary optic atrophy type 3 is caused by mutation in the OPA3 gene with cytogenetic location 19q13.32. The mutation in this gene is associated with childhood-onset vision loss with cataract. It can also be associated with type III methylglutaconic aciduria.
Leber hereditary optic neuropathy results from mitochondrial point mutations in mtDNA 11778G>A, 14484T>C, or 3460G>A mutations.
Consecutive atrophy
Consecutive atrophy is an ascending type of atrophy (eg, chorioretinitis, pigmentary retinal dystrophy, cerebromacular degeneration) that usually follows diseases of the choroid or the retina.
Circulatory atrophy (vascular)
Circulatory atrophy is an ischemic optic neuropathy observed when the perfusion pressure of the ciliary body falls below the intraocular pressure. Circulatory atrophy is observed in central retinal artery occlusion, carotid artery occlusion, and cranial arteritis.
Metabolic atrophy
It is observed in disorders such as thyroid ophthalmopathy, juvenile diabetes mellitus, nutritional amblyopia, toxic amblyopia, tobacco, methyl alcohol, and drugs (eg, ethambutol, sulphonamides).
Demyelinating atrophy
It is observed in diseases such as multiple sclerosis and Devic disease.
Pressure or traction atrophy
It is observed in diseases such as glaucoma and papilledema.
Postinflammatory atrophy
It is observed in diseases such as optic neuritis, perineuritis secondary to inflammation of the meninges, and sinus and orbital cellulites.
Traumatic optic neuropathy
The exact pathophysiology of traumatic optic neuropathy is poorly understood, although optic nerve avulsion and transection, optic nerve sheath hematoma, and optic nerve impingement from a penetrating foreign body or bony fragment all reflect traumatic forms of optic nerve dysfunction that can lead to optic atrophy. [1]
Regardless of etiology, optic atrophy is associated with variable degrees of visual dysfunction, which may be detected by one or all of the optic nerve function tests. [1]
See Other Tests.
Radiation optic neuropathy
Radiation optic neuropathy more frequently occurs with radiation doses of at least 5,000 centigray. It may be result from radiation damage to the optic nerve vasculature or the optic nerve parenchyma itself.
Epidemiology
Frequency
United States
According to Tielsch et al, the prevalence of blindness attributable to optic atrophy was 0.8%. [9]
The prevalence of visual impairment and blindness attributable to optic atrophy was found to be 0.04% and 0.12%, respectively. [10]
International
Optic atrophy type 1 is believed to affect approximately 1 in 35,000 individuals globally. However, it is more prevalent in Denmark, affecting about 1 in 10,000 people.
Mortality/Morbidity
Optic atrophy is not a disease but an end outcome; thus, morbidity and mortality in optic atrophy depend on the etiology.
Race
Optic atrophy is more prevalent in African Americans (0.3%) than in Whites (0.05%).
Sex
There is no sexual predisposition noted.
Age
Optic atrophy is seen in any age group.
Prognosis
Optic atrophy is an end-stage disease, although anecdotal reports have described significant vision improvement in the fellow eye following acute optic neuropathy or geographic atrophy. [11, 12] Early and intensive treatment in nutritional optic neuropathy can provide patients with near-normal vision.
RNFL (retinal nerve fiber layer) OCT measurements have revealed marked optic nerve axon reserve before symptomatic presentation, after which small changes in nerve fiber loss lead to significant decrease in vision. [13] Therefore, one should remember early identification and treatment of cause is the key to save useful vision.
-
Normal optic nerve histopathology.
-
Glaucomatous optic atrophy histopathology.
-
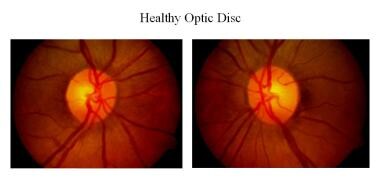
Healthy optic disc.
-
Nonarteritic anterior ischemic optic neuropathy.
-
Arteritic anterior ischemic optic neuropathy, cilioretinal artery occlusion.
-
Primary optic atrophy.
-
Optic atrophy following papilledema (secondary).
-
Glaucomatous optic atrophy.
-
Juvenile open-angle glaucoma (JOAG) with optic pallor.
-
Consecutive optic atrophy following panretinal photocoagulation (PRP).
Tables
|
Postneuritis |
Ischemic Arteritic |
Ischemic Nonarteritic |
Compressive |
Age |
15-50 y |
Approximately 70 y |
Sixth decade |
Varies based on cause |
Sex |
Multiple sclerosis F>M |
F>M |
F=M |
Varies based on cause |
Visual acuity |
Varies from mild blurring (34%) and moderate loss of acuity (12%) to severe or total loss of light perception (complete blindness) in 54% of cases, to no light perception. The loss of vision is acute and progressive.--Vision usually recovers within 2 mo |
< 20/200 (6/60) |
>20/200 (6/60) |
Varies from mild blurring to no light perception |
Color vision |
Color vision > vision loss |
Color vision loss = vision loss |
Color vision loss = vision loss |
Color vision = vision loss |
RAPD* |
+ |
+ |
+ |
+ |
Motility |
Painful movement in cases of retrobulbar neuritis |
Normal |
Normal |
Depends on the site of compression |
Nystagmus |
In multiple sclerosis, vertical nystagmus (upbeating or downbeating) may be seen |
No |
No |
See-saw nystagmus in optic chiasm compression |
Optic disc |
Temporal pallor |
Pallid disc edema |
Segmental disc edema |
Bow-tie pallor seen in optic chiasm compression; varies in other instances |
Electrophysiologic study |
VEP-increased latency < †> |
VEP-reduced amplitude |
VEP-reduced amplitude |
Reduced VEP amplitude |
Neuroimaging (CT, MRI) |
In multiple sclerosis, hyperechoic lesions are seen in the brain on MRI |
- |
- |
For exact location of compression |
Other associations |
|
Headache, scalp tenderness, jaw claudication |
Hypertension and diabetes |
Headache, vomiting, and focal neurologic deficits |
*RAPD - Relative afferent pupil defect < †>VEP - Visual-evoked potential |
||||

What would you like to print?
- Italian Duchenne Muscular Dystrophy Drug Gets OK in Europe
- Faster Brain Atrophy Linked to MCI
- Vaginal Gel Has Long-Term Benefit for Menopause-Related Vaginal Atrophy
-
 Could This Child Have Spinal Muscular Atrophy?
Could This Child Have Spinal Muscular Atrophy?
-
Q&A: Spinal Muscular Atrophy Treatment Update
-
Spinal Muscular Atrophy: 5 Things to Know






