Background
Hyperinsulinism (HI), in which the pancreatic beta cells secrete an excessive amount of insulin, is the most common cause of severe, persistent hypoglycemia in infants and children. [1] [2] Infants with uncontrolled hypoglycemia caused by HI are at risk for seizures or permanent brain damage. There are two forms of HI, as follows:
-
Transient neonatal HI - Develops soon after birth and generally resolves by age 6 months
-
Persistent HI - Has a more protracted duration; in some cases, the symptoms may manifest after infancy
Transient HI is temporary, but it can cause brain damage if left untreated. Risk factors for this form of HI include the following:
-
Maternal diabetes
-
Perinatal stress
-
Maternal perinatal hypertension
-
Intrauterine growth restriction or large for gestational age (LGA)
The causes of persistent HI are largely genetic. [2] [3] [4, 5] Mutations in more than 30 genes that play a role in regulating beta-cell insulin secretion have been implicated in the pathogenesis of HI. [6, 7] Genetic forms are sometimes classified into seven subtypes [8] :
-
KATP-hyperinsulinism
-
GDH-hyperinsulinism
-
GK-hyperinsulinism
-
SCHAD-hyperinsulinism
-
UCP2-hyperinsulinism
-
HNF4A and HNF1A-hyperinsulinism
-
MCT1-hyperinsulinism
Several syndromic genetic forms of HI have also been identified (eg, Beckwith-Wiedemann, Kabuki, and Turner syndromes). [6]
Maternal diet apparently does not have a significant effect on neonatal cord blood insulin, C-peptide, or plasma glucose levels, although a lower maternal glycemic load appears to be associated with lower adiposity in infants born to these women. [9]
There remain approximately 50% of diazoxide-responsive cases and 10% of diazoxide-unresponsive cases of persistent HI with unknown etiology, suggesting that additional genes may be identified in the pathogenesis of HI. [8] (Diazoxide is the first-line drug for controlling hypoglycemia in HI,)
Diagnostic criteria include plasma glucose levels of less than 3 mmol/L with detectable serum insulin and C-peptide, low serum ketone bodies, and low serum fatty acids. An intravenous glucose infusion rate greater than 8 mg/kg/min (normally, 4-6 mg/kg/min) strongly supports the diagnosis. [10]
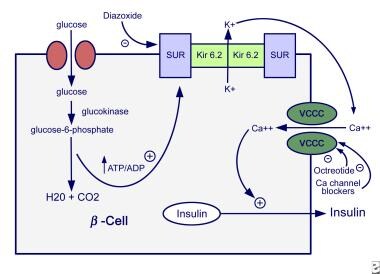
Diazoxide is ineffective in some genetic forms of HI (KATP-HI, GK-HI). Octreotide may be used in diazoxide-unresponsive patients but is often ineffective because of down-regulation of the somatostatin receptor, and it carries a risk of causing necrotizing enterocolitis and death. [6] Severe cases of congenital HI may be unresponsive to either diazoxide or octreotide, requiring intensive management with tube feedings, near-total pancreatectomy, or partial pancreatectomy. The image below illustrates the mechanisms of insulin secretion.
 Mechanisms of insulin secretion.
Mechanisms of insulin secretion.
Pathophysiology
The differential diagnosis of hypoglycemia is extensive, and determining the underlying cause is often difficult. An understanding of glucose homeostasis can help to narrow the differential diagnosis. In the fasting state, glucose is provided through glycogenolysis in the liver. After a few hours of fasting, insulin levels fall, and increased lipolysis creates free fatty acids and glycerol. Fatty acids do not cross the blood-brain barrier and, therefore, are not used by the brain. However, fatty acids are used by the heart and muscle. Increased free fatty acids result in the production of ketones, and the brain is able to metabolize ketones as an alternative source of fuel.
Disorders that result from defective glycogenolysis in the liver lead to hypoglycemia within a few hours of fasting. This hypoglycemia occurs in the setting of low insulin levels.
Disorders of fat metabolism result in the unavailability of free fatty acids and ketones as alternative fuels. Hypoglycemia occurs after several hours of fasting. Circulating insulin levels also are low.
Growth hormone deficiency and hypocortisolemia also can cause hypoglycemia associated with low insulin levels, possibly by unopposed insulin action and decreased ketogenesis.
Hypoglycemia associated with elevated insulin levels makes certain disorders unlikely, such as defects in gluconeogenesis, free fatty acid synthesis, and ketogenesis; growth hormone deficiency; and cortisol deficiency. Conversely, hypoglycemia associated with ketonuria makes HI less likely.
Glucose and several amino acids stimulate insulin secretion under physiologic conditions, and the sequence of events leading to insulin secretion is well delineated. The rate of insulin secretion is dependent on the ratio of adenosine triphosphate (ATP) to adenosine diphosphate (ADP) within the beta cell. The rate of glucose entry into the beta cell is facilitated by a glucose transporter, and the entry rate exceeds the oxidation rate of glucose. Glucokinase is the rate-limiting step of glycolysis (ATP production), not glucose transport.
The first step in glycolysis (ie, conversion of glucose to glucose-6-phosphate [G6P] by glucokinase) is the rate-limiting step in glucose metabolism. Thus, glucokinase regulates the rate of glucose oxidation and subsequent insulin secretion. An increase in the intracellular ATP/ADP ratio activates ATP-sensitive potassium-dependent (KATP) channels in the cell membrane. Subunits of the KATP channel include the sulfonylurea receptor (SUR1, encoded by the gene ABCC8) and the potassium inward rectifier channel (Kir6.2, encoded by the gene KCNJ11). Activation leads to closure of the potassium channel and depolarization of the cell membrane. Opening of a voltage-gated calcium channel allows influx of calcium and results in insulin secretion.
Transient HI usually results from environmental factors such as maternal diabetes and birth asphyxia. However, children with persistent HI may have a genetic defect that results in inappropriate secretion of insulin.
Etiology
Transient causes of HI
Infants of mothers with diabetes
During gestation, glucose is freely transferred across the placenta. Prolonged hyperglycemia in poorly controlled maternal diabetes results in fetal hyperglycemia. Fetal hyperglycemia induces fetal pancreatic beta-cell hyperplasia with resultant hyperinsulinemia and macrosomia. Withdrawal of the transplacental supply of glucose after birth leads to a precipitous drop in the concentration of glucose. When neonates present with signs and symptoms of hypoglycemia, many require infusion of large quantities of glucose to maintain normal blood glucose levels. HI typically resolves within 1-2 days following birth (see Infant of Diabetic Mother).
Prolonged HI in infants who are SGA and asphyxiated newborns
Infants who are small for gestational age (SGA), experience maternal toxemia, or have birth asphyxia are at increased risk for developing hypoglycemia. These infants have high rates of glucose metabolism and may require dextrose infusions of as high as 20 mg/kg/min to maintain euglycemia. Some evidence suggests that this may be due to hyperinsulinemia, although the exact mechanisms are still unclear. These patients may have prolonged hypoglycemia for as long as 2-4 weeks following birth. Afterward, the hypoglycemia appears to resolve completely.
Erythroblastosis fetalis
Neonates with severe Rh isoimmunization have islet cell hyperplasia and HI. The cause of HI is unknown. Researchers hypothesize that elevated levels of glutathione from massive hemolysis may serve as a stimulus for insulin release.
Drug-induced causes of HI
Surreptitious insulin administration
This phenomenon is rare but may occur in the setting of Munchausen syndrome by proxy. The timing of hypoglycemia is unpredictable and occurs when the offender has access to the patient. Laboratory evaluation reveals elevated insulin levels and a low serum C-peptide level.
Ingestion of oral hypoglycemic agents
Toddlers may accidentally ingest drugs prescribed for adults with diabetes (eg, sulfonylureas). Depending on the half-life of the preparation ingested, the duration of hypoglycemia varies. Glucose infusion (to maintain normoglycemia) is the treatment of choice. On rare occasions, diazoxide may be needed to suppress insulin secretion.
Blood transfusion
Certain preparations of blood products (eg, citrated blood) have large amounts of dextrose. During transfusion, the high glucose load triggers insulin secretion. Problems arise when the transfusion is completed. Elevated insulin levels could lead to a precipitous drop in blood glucose levels. This fall typically occurs about 2 hours after transfusion.
Malposition of the umbilical artery catheter
Malposition of the umbilical artery catheter in neonates may be associated with hypoglycemia and hyperinsulinemia. Repositioning of the catheter usually resolves the hypoglycemia and hyperinsulinemia. Theoretically, this problem may be caused by a high glucose load administered to the celiac axis. Localized hyperglycemia would induce insulin secretion and result in hypoglycemia in the systemic circulation.
Congenital causes
Beckwith-Wiedemann syndrome, a congenital cause of HI, includes symptoms of omphalocele, macroglossia, and visceromegaly. Infants with this syndrome have generalized islet cell hyperplasia
Hyperinsulinemic hypoglycemia may be difficult to control. These patients require large quantities of glucose. Treatment with diazoxide is often needed to control hyperinsulinemia. HI usually spontaneously resolves when the infant is aged several weeks or months.
Focal causes of HI
Patients with congenital HI may demonstrate focal histologic abnormalities, which most pathologists label as islet adenomatosis or beta-cell adenoma. As patients present with hyperinsulinemic hypoglycemia at older ages (>1 yr), they are increasingly more likely to have the focal form of HI.
A study that used preoperative pancreatic catheterization and intraoperative histologic studies suggested that as many as half of all neonates presenting with congenital HI have focal islet cell hyperplasia. Focal causes of HI can be treated, and possibly cured, with partial pancreatectomy.
Genetic causes
Pancreatic B-cell KATP-channel defects
Recessive mutations on chromosome 11 lead to alterations in the potassium channel on the plasma membrane of pancreatic beta cells.
Mutations in the ABCC8 and KCNJ11 genes create a nonfunctional potassium channel with membrane depolarization and unchecked insulin secretion. Mutations of the ABCC8 gene are more common than mutations of the KCNJ11 gene; ABCC8 mutations have been found more frequently in the less heterogeneous populations of Saudi Arabia and Ashkenazi Jews.
Patients with the autosomal recessive disorder present with high birth weights from the anabolic effects of insulin in utero. These disorders cannot be controlled with diazoxide, which activates SUR1 to suppress insulin secretion. Thus, pancreatectomy is often required. For this subset of patients, near-total pancreatectomy achieves the best glycemic control during infancy.
GCK (encoding glucokinase) mutations
Mutations of the GCK gene can be autosomal dominant or recessive. The GCK mutations increase the affinity of glucokinase for glucose (ie, lower intrinsic Km for the glucose binding site). Accelerated rates of glycolysis result in an increased ATP/ADP ratio and increased insulin secretion. Patients with these mutations have a milder form of HI than patients with potassium channel defects. These patients also respond well to diazoxide treatment. In some patients, treatment can be discontinued after several years.
Hyperinsulinism-hyperammonemia (HH) syndrome due to GLUD1 mutation
GLUD1 encodes glutamate dehydrogenase, and mutations reported in this gene to date have been transmitted in an autosomal dominant inheritance. Early in infancy, patients with this genetic mutation present with hypoglycemic seizures, which are unrelated to the hyperammonemia per se. Delays in diagnosis and definitive care are unfortunately common because of the rarity of this disease.
GLUD1 mutation affects both hepatocytic and islet function. Two metabolic pathways use glutamate dehydrogenase: leucine glutamate dehydrogenase–mediated oxidation in beta cells produces ATP, which induces insulin release; glutamate dehydrogenase also reduces intrahepatocytic glutamate concentration, and glutamate depletion down-regulates the first step of the urea cycle to convert ammonium to urea.
Excessive activity of glutamate dehydrogenase thus increases the rate of insulin release by beta cells in the pancreas and impairs the detoxification of ammonia by hepatocytes in the liver.
Patients with one of these GLUD1 gene mutations present with low blood glucose levels and persistent, mild elevations of serum ammonia to 100-200 µmol/L. This hyperammonemia is not affected by fasting, intravenous L-leucine challenge, oral L-leucine challenge, or glycemic control by medication. Indeed, the hyperammonemia itself has not been associated with clinical consequences, in contrast to the hypoglycemia, which can cause permanent brain damage. GLUD1 mutations tend to display less severe hypoglycemia and respond to diazoxide.
Exercise-induced HI (EIHI)
EIHI is characterized by inappropriate insulin secretion that leads to hypoglycemia during exercise. Promoter-activating mutations of the SLC16A1 gene encode a monocarboxylate transporter (MCT1) that mediates the movement of lactate and pyruvate across cell membranes and causes anaerobic exercise–induced hypoglycemia as a dominantly inherited trait. Patients typically become hypoglycemic 30-45 minutes after a period of intensive exercise.
Uncoupling protein 2 (UCP2)
Loss-of-function mutations encoding UCP2 lead to an increased ATP synthesis and enhanced glucose-stimulated insulin secretion. Diazoxide responsive, this rare form of HH syndrome is thought to be transient.
3-Hydroxyacyl-CoA dehydrogenase (HADH)
HADH is inherited as an autosomal recessive manner. Diazoxide responsive, this rare disorder is characterized by increased levels of 3-hydroxybutyryl-carnitine in blood and 3-hydroxyglutaric acid in urine. However, the precise mechanism of HI in patients with an HADH deficiency is not well understood.
Hepatocyte nuclear factor 4 alpha (HNF-4A)
HNF-4A, encoded by the HNF4A gene, is a transcription factor that plays an important role in pancreatic development, maintenance of B-cell mass, and regulation of insulin secretion. HNF4A gene mutations can cause increased birth weight, macrosomia, and transient HH syndrome in the neonatal period, with evolution to decreased insulin secretion and maturity-onset diabetes of the young type 1 (MODY1) later in life. [11]
Epidemiology
Transient HI is relatively common in neonates. An infant of a diabetic mother, an infant who is small or large for gestational age, or any infant who has experienced severe stress may have high insulin concentrations. In contrast, congenital HI is rare.
In a literature review, Lapidus et al estimated the international prevalence of persistent HI in populations of European ancestry to be 3.5 per 100,000 births. However, the investigators cautioned that this figure is a minimum estimate, being based on data derived only from patients whose diagnosis involved genetic testing. [12]
In the United States, HI is estimated to occur in 1 in 30,000-50,000 live births. [8] Internationally, the estimated incidence of congenital HI runs from 1 in 50,000 births in Holland to 1 in 2500 in Saudi Arabia (with the latter incidence owing to high rates of consanguinity). The incidence in Ashkenazi Jews, as estimated using the carrier frequency for two recessive KATP-HI founder mutations, is 1 in 10,816. [6] In Japan, the incidence of transient neonatal HI is estimated at 1 in 17,000 births and that of persistent hypoglycemia at 1 in 35,400 births. [13]
Prognosis
Multiple factors affect prognosis, such as the severity of the disease at presentation, duration of hypoglycemia, etiology of HI, and presence of neurologic complications. Owing to diagnostic delays and inadequate treatment, infants with HI have a risk of permanent brain damage as high as 25-50%. [6]
Unfortunately, many infants with HI remain undiagnosed, misdiagnosed, or inadequately treated for several months before definitive management.
Improvements in diagnostic techniques will make earlier and more appropriate surgical intervention (partial pancreatectomy or near-total pancreatectomy) possible. However, patients who have had near-total pancreatectomy are at risk for developing exocrine pancreatic insufficiency and diabetes mellitus. [14]
Diabetes mellitus, which develops in patients with diffuse disease, is caused by dysregulation of insulin secretion in the residual beta cells after pancreatectomy.
Complications
Complications of congenital HI include seizures, developmental delays, and death.
In a retrospective chart review, symptomatic hypertrophic cardiomyopathy (HCM) was found in approximately 15% (95% CI, 6-23%) of infants with congenital HI (95% CI, 6-23%). All of the affected infants had the KATP-channel form of HI and ultimately failed medical management, requiring pancreatectomy. The researchers noted that echocardiography was performed only on symptomatic children, so that the incidence of cardiomyopathy in infants with congenital HI is likely higher. Additionally, HCM was identified only in patients who had undergone pancreatectomy, suggesting that infants with more severe HI have a higher risk of a disturbance in cardiomyocyte growth. [15]
Patient Education
Counsel the patient, family members, and school personnel on how to recognize the symptoms of hypoglycemia and how to administer glucose in the event of a hypoglycemic episode. Prolonged fasting should be avoided. Seek medical attention if emesis or anorexia develops. Families should be equipped with glucagon and instructed in its use in case hypoglycemia does occur.
-
Mechanisms of insulin secretion.
Tables

What would you like to print?
- Can Tirzepatide Put Type 2 Diabetes Into Remission?
- How Much Exercise Is Advised for Patients With Type 2 Diabetes?
- What's the Goal in Treating Type 2 Diabetes?
-
 Choosing Which Agents to Use in the Management of CKD in Type 2 Diabetes Patients: SGLT2 Inhibitors vs GLP-1s
Choosing Which Agents to Use in the Management of CKD in Type 2 Diabetes Patients: SGLT2 Inhibitors vs GLP-1s
- Anticoagulation and Antiplatelet Therapy for Atrial Fibrillation and Stable Coronary Disease
-
Heart Failure, Incretin Therapies, and Type 2 Diabetes


 Complications of Diabetes
Complications of Diabetes



