Practice Essentials
Xeroderma pigmentosum is a rare disorder transmitted in an autosomal recessive manner. It is characterized by acute photosensitivity, pigmentary changes, premature skin aging, and malignant tumor development. Early signs include freckle-like pigmentation of the face, appearing before age two years (see the image below). The disease results from mutations in genes involved in DNA repair, which leads to a cellular hypersensitivity to ultraviolet (UV) radiation. Affected individuals are at markedly increased risk of sunlight-induced skin cancers at a young age, as well as extracutaneous cancers. [1, 2]
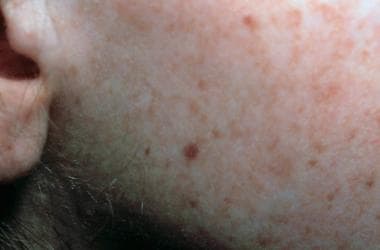 Face of a toddler with xeroderma pigmentosum, representative of an early stage of the disease. Note the freckling and the scaling. Courtesy of Neil S. Prose, MD, Duke University Medical Center, Durham, North Carolina.
Face of a toddler with xeroderma pigmentosum, representative of an early stage of the disease. Note the freckling and the scaling. Courtesy of Neil S. Prose, MD, Duke University Medical Center, Durham, North Carolina.
Most individuals with xeroderma pigmentosum also have ocular involvement, with conjunctival inflammation, corneal lesions, and eyelid pathology. [3, 4] Some experience neurologic degeneration (eg, progressive hearing loss, cognitive impairment, ataxia). [1, 5]
Background
Xeroderma pigmentosum (XP) was first described in 1874 by Hebra and Kaposi. In 1882, Kaposi coined the term xeroderma pigmentosum for the condition, referring to its characteristic dry, pigmented skin. [2] Albert Neisser first described neurologic abnormalities associated with xeroderma pigmentosum in 1883. Cleaver's seminal work in 1968 elucidated the pathophysiology of xeroderma pigmentosum by demonstrating defective DNA repair. Further studies of this defect led to significant progress in the understanding of nucleotide excision repair (NER) mechanisms under normal and pathologic conditions. [6]
Pathophysiology
The basic defect in xeroderma pigmentosum is in nucleotide excision repair (NER), leading to deficient repair of DNA damaged by UV radiation. [7] This extensively studied process consists of the removal and the replacement of damaged DNA with new DNA. Two types of NER exist: global genome (GG-NER) and transcription coupled (TC-NER). [8] The last decade has seen the cloning of the key elements of NER, and the process has been reconstituted in vitro.
Seven xeroderma pigmentosum repair genes, XPA through XPG, have been identified. [5] These genes play key roles in GG-NER and TC-NER. Both forms of NER include a damage-sensing phase, performed in GG-NER by the product of the XPC gene complexed to another factor. In addition, the XPA gene product has been reported to have an affinity for damaged DNA. Therefore, XPA likely also plays a role in the damage-sensing phase.
Following detection of DNA damage, an open complex is formed around the damaged area. The XPG gene product is required for the open complex formation. The XPB and XPD gene products are part of a 9-subunit protein complex (TFIIH) that is also needed for the open complex formation. Subsequently, the damaged DNA is removed. The XPG and XPF genes encode endonucleases; however, the XPF gene product functions as an endonuclease when complexed to another protein. The resulting gap is filled in with new DNA by the action of polymerases.
A xeroderma pigmentosum variant has also been described. The defect in this condition is not in NER, but is instead in postreplication repair. In the xeroderma pigmentosum variant, a mutation occurs in DNA polymerase η. [9, 10]
Seven complementation groups, XPA through XPG, corresponding to defects in the corresponding gene products of XPA through XPG genes, have been described. These entities occur with different frequencies (eg, XPA is relatively common, whereas XPE is fairly rare), and they differ with respect to disease severity (eg, XPG is severe, whereas XPF is mild) and clinical features. Cockayne syndrome can rarely occur with XPB, XPD, and XPG. [11]
The continued presence of repair proteins at sites of DNA damage may also contribute to the pathogenesis of cutaneous cancer, as has been shown in XPD. [12]
In addition to the defects in the repair genes, the pathogenesis of xeroderma pigmentosum may involve immunosuppressive effects of UV-B radiation. [13] Although typical symptoms of immune deficiency, such as multiple infections, are not usually observed in patients with xeroderma pigmentosum, several immunologic abnormalities have been described in the skin of patients with xeroderma pigmentosum. Clinical studies of the skin of patients with xeroderma pigmentosum indicate prominent depletion of Langerhans cells induced by UV radiation.
Various other defects in cell-mediated immunity have been reported in xeroderma pigmentosum. These defects include impaired cutaneous responses to recall antigens, decreased ratio of circulating T-helper cells to suppressor cells, impaired lymphocyte proliferative responses to mitogen, impaired production of interferon in lymphocytes, and reduced natural killer cell activity.
In addition to their role in DNA repair, xeroderma pigmentosum proteins also have other functions. For example, Fréchet et al [14] have shown that matrix metalloproteinase 1 is overexpressed in dermal fibroblasts from patients with XPC. [15] They also demonstrated accumulation of reactive oxygen species in these fibroblasts in the absence of exposure to UV. They concluded that the XPC protein has roles in addition to NER. Matrix metalloproteinase 1 overexpression has been shown to occur in both aging of skin and carcinogenesis.
XPG has been shown to form a stable complex with the transcription factor TFIIH, as mentioned above. Some manifestations of XPG/Cockayne syndrome in patients may therefore be due to abnormal transcription. [16]
With respect to neurodegeneration seen in some cases of xeroderma pigmentosum, it may be associated with TC-NER rather than GG-NER. [17]
Epidemiology
Frequency
United States
The frequency in the United States is approximately 1 case per 250,000 population. Group XPC is the most common form in the United States.
International
The frequency in Europe is approximately 1 case per 250,000 population. In Japan, it is higher, 1 case per 40,000 population. [18] Groups XPA and XPC are the most common. Group XPA is the most common form in Japan. [19]
Race-, sex-, and age-related demographics
Note the following:
-
Cases of xeroderma pigmentosum are reported in persons of all races.
-
An equal prevalence has been reported in males and females.
-
The disease is usually detected at age 1-2 years.
Prognosis
Individuals with xeroderma pigmentosum have a shortened lifespan: Less than 40% survive beyond age 20 years, although individuals with milder disease may survive beyond middle age. Individuals with this disease develop multiple cutaneous neoplasms at a young age. Two important causes of mortality are metastatic malignant melanoma and squamous cell carcinoma. [20] Patients younger than 20 years have a 1000-fold increase in the incidence of nonmelanoma skin cancer and melanoma. The mean age of skin cancer development is 8 years in patients with xeroderma pigmentosum, compared with 60 years in the healthy population. Actinic damage occurs between ages 1 and 2 years.
Patient Education
Constant education of the patient is the most important objective in the management of xeroderma pigmentosum. The need for adequate solar protection cannot be overemphasized and should be reinforced at every visit.
Sunblocks should be used, even in winter months and during evening and early morning hours. The exposed surfaces of the skin should be shielded with protective, double-layered clothing and broad-brimmed hats. The eyes should be shielded with UV-absorbing sunglasses with side shields.
Even unlikely sources of illumination can prove hazardous and should be pointed out to patients; for example, fluorescent lights that emit radiation below 320 nm can be dangerous.
The Xeroderma Pigmentosum Society provides information for affected individuals, their families, and the public. It also provides peer support for patients and their families.
-
Face of a toddler with xeroderma pigmentosum, representative of an early stage of the disease. Note the freckling and the scaling. Courtesy of Neil S. Prose, MD, Duke University Medical Center, Durham, North Carolina.
-
Back of an adolescent with xeroderma pigmentosum, representing a later stage of the disease. Note the mottled hyperpigmentation and atrophy. Courtesy of Neil S. Prose, MD, Duke University Medical Center, Durham, North Carolina.
-
Histologic features of actinic keratosis in an individual with xeroderma pigmentosum. Note the atypia of the keratinocytes and the parakeratosis.
-
Sunlight-induced dermatologic abnormalities in a patient with xeroderma pigmentosum.
-
Typical skin manifestation of xeroderma pigmentosum with numerous areas of hypopigmentation and freckles (ie, solar lentigines) with different intensities of pigmentation.
-
Axial T2-weighted MRI of the brain of a 47-year-old woman with xeroderma pigmentosum, complementary group D. She developed progressive ataxia and dementia at age 44 years. Note severe atrophy and normal signal from the white matter.
-
Coronal T1-weighted MRI image of the brain of a 47-year-old woman who developed progressive ataxia and dementia at age 44 and was found to have xeroderma pigmentosum, complementary group D (see image above). Severe diffuse atrophy involves the brain stem and cerebellum.
