Practice Essentials
Keratoacanthoma (KA) is a relatively common low-grade tumor that originates in the pilosebaceous glands. KA is a subtype of squamous cell carcinoma (SCC) with a tendency toward spontaneous regression. [1, 2, 3] Dermatopathologists refer to the lesion as squamous cell carcinoma, keratoacanthoma type (SCC–KA type). However, some have argued for a distinction between keratoacanthoma and SCC based on gene expression [4] or a cutaneous marker. [5]
Keratoacanthoma is characterized by rapid growth over a few weeks to months, followed by spontaneous resolution over 4-6 months in most cases. Keratoacanthoma may progress rarely to invasive or metastatic carcinoma. Whether such cases were initially SCC or keratoacanthoma, the reports highlight the difficulty of distinctly classifying individual cases. [6, 7, 8, 9]
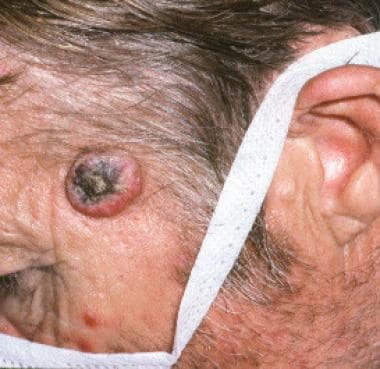
The image below depicts keratoacanthoma of the left forehead.
Pathophysiology
Trauma/local injury, ultraviolet light, chemical carcinogens, human papillomavirus, genetic factors (mutations in p53 or H-Ras), and immunocompromised status have been implicated as etiologic or triggering factors. Cancer immunotherapies, such as immune checkpoint inhibitors, can have cutaneous adverse effects, and the flourishing of these targeted therapies has led to a marked increase in keratoacanthoma incidence. [10, 11]
Keratoacanthoma is associated with syndromes such as Muir-Torre syndrome, Ferguson-Smith syndrome, xeroderma pigmentosum, and incontinentia pigmenti. A rare disorder, generalized eruptive keratoacanthoma of Grzybowski, is discussed in further detail in DDx.
Keratoacanthoma and conventional SCC share similar epidemiologic features, which suggests a possible common pathogenesis such as actinic damage. [12] In population-based studies in Kauai, Hawaii, similarities between keratoacanthoma and SCC included the following:
-
Anatomic site – Most keratoacanthomas and SCCs developed on head/neck and limbs (keratoacanthoma, 78%; SCC, 85%)
-
Patient age – The incidence of keratoacanthoma and SCC increased significantly after age 64 years; the average age of patients was 67 years in keratoacanthoma and 66 years in SCC [12]
-
Male predominance – Male-to-female ratios for both conditions were 2:1 [12]
Etiology
The definitive cause of keratoacanthoma remains unclear; however, several potentiating factors should be considered. Epidemiologic data on keratoacanthoma are notably similar to SCC and Bowen disease (SCC in situ) concerning age, sex, and the anatomic site of lesions. These data strongly support a common etiology among keratoacanthoma, SCC, and Bowen disease. Epidemiologic data support ultraviolet light as an important etiologic factor.
Industrial workers exposed to pitch and tar have been well established as having a higher incidence of keratoacanthoma, as well as SCC. [14] Additionally, a 2006 study suggested a strong association between cigarette smoking and the development of keratoacanthoma. [15]
Trauma (iatrogenic or non-iatrogenic), human papillomavirus infection (specifically with types 9, 11, 13, 16, 18, 24, 25, 33, 37, and 57), [16, 17] genetic factors, and immunocompromise also have been implicated as etiologic factors.
Merkel cell polyomavirus does not play a pathogenic role in keratoacanthoma. [18]
Twenty percent of patients who had metastatic melanoma and were treated with vemurafenib, a novel BRAF V600E inhibitor, may develop eruptive keratoacanthoma or squamous cell carcinoma. [19]
Finally, research has identified that up to one third of keratoacanthomas harbor chromosomal aberrations. Recurrent aberrations include gains on 8q, 1p, and 9q with deletions on 3p, 9p, 19p, and 19q. One other report identified a 46,XY,t(2;8)(p13;p23) chromosomal aberration. [20, 21, 22, 23]
Epidemiology
Frequency
United States
The sole published study on keratoacanthoma in a White US population took place in Hawaii and estimated the incidence at 106 cases per 100,000. This study reported keratoacanthoma incidence equal to SCC and challenged the commonly reported incidence ratio of keratoacanthoma to SCC of 1:3. [12, 13]
Race-, sex-, and age-based demographics
Based on the Hawaiian data, the incidence of keratoacanthoma in ethnic Japanese, Filipino, and Hawaiian populations has been estimated to be 22, 7, and 6 cases per 100,000 population, respectively—approximately one fifth to one sixteenth of the incidence rate found in US Whites. In other studies, the ratio of keratoacanthoma to SCC has ranged from 1:0.6 to 1:5 in different geographic locations. [12, 24, 25, 26]
Keratoacanthoma is less common in darker-skinned individuals. [12, 24, 25, 26]
The male-to-female ratio for keratoacanthoma is 2:1.
Keratoacanthoma has been reported in all age groups, but incidence increases with age. Keratoacanthoma is rare in persons younger than 20 years. Peak incidence occurs in the seventh decade or beyond.
Prognosis
The prognosis for keratoacanthoma is excellent following excisional surgery. Recurrent tumors may require more aggressive therapy. It is important to follow patients with a history of keratoacanthoma for development of new primary skin cancers (SCC in particular). The reclassification of keratoacanthoma as SCC–KA type reflects the difficulty in histologic differentiation.
Uncommonly, keratoacanthomas may exhibit an aggressive growth pattern. Keratoacanthoma infrequently presents as multiple tumors and may enlarge (5-15 cm), become aggressive locally, or rarely, metastasize. [27, 28]
-
Keratoacanthoma (squamous cell carcinoma-keratoacanthoma or SCC-KA type) on inner canthus.
-
Keratoacanthoma of the left forehead.
-
Close-up view of the keratoacanthoma.
-
Keratoacanthoma lesion (squamous cell carcinoma-keratoacanthoma or SCC-KA type).
Tables

What would you like to print?
- Darker Skin Tones Underrepresented on Skin Cancer Education Websites
- Rising Skin Cancer Rates: Epidemic or Overdiagnosis?
- 'Superclinic' Powers Through Skin Cancer Referrals
-
 A Surgeon's Guide to Hidradenitis Suppurativa
A Surgeon's Guide to Hidradenitis Suppurativa
-
Postpartum Thyroiditis: Risk Factors, Workup, and Management
-
Recognizing and Treating Hyperprolactinemia



 Deadly Skin Cancers
Deadly Skin Cancers