Practice Essentials
The term pemphigus (from Greek pemphix "bubble or blister") describes a group of autoimmune blistering diseases of the skin and mucous membranes characterized histologically by intraepidermal blister and immunopathologically by the finding of in-vivo bound and circulating immunoglobulin G (IgG) antibody directed against the cell surface of keratinocytes. Pemphigus was once considered to include most bullous eruptions of the skin, but diagnostic tests have improved, and bullous diseases have been reclassified accordingly.
The three primary subsets of pemphigus, each of which has its own distinct clinical and immunopathologic features, are as follows [1] :
-
Pemphigus vulgaris
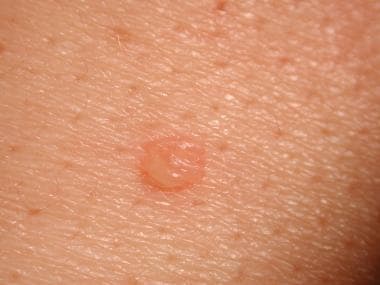
Pemphigus vulgaris (see the image below) accounts for approximately 70% of pemphigus cases. It is a potentially life-threatening disease, with a mortality of approximately 5-15%. [2]
Signs and symptoms
Mucous membranes
Mucous membranes of the oral cavity are involved in almost all patients with pemphigus vulgaris. Patients may have ill-defined, irregularly shaped, gingival, buccal, or palatine erosions, which are painful and slow to heal. Intact bullae are rare in the mouth. Erosions may be seen on any part of the oral cavity, and they may spread to involve the larynx, with subsequent hoarseness. In juvenile pemphigus vulgaris, stomatitis is the presenting complaint in more than 50% of cases.
Other mucosal surfaces may be involved, including the conjunctiva, esophagus (causing odynophagia and/or dysphagia), labia, vagina, cervix, vulva, penis, urethra, nasal mucosa, and anus.
Skin
The primary lesion of pemphigus vulgaris is a flaccid blister filled with clear fluid that arises on healthy skin or on an erythematous base. Blisters are fragile and may rupture, producing painful erosions (the most common skin presentation).
Nails
Acute or chronic paronychia, subungual hematomas, and nail dystrophies affecting one or several fingers or toes have been reported with pemphigus vulgaris.
Vegetating pemphigus vulgaris
Lesions in skin folds readily form vegetating granulations. In some patients, erosions tend to develop excessive granulation tissue and crusting; these individuals display more vegetating lesions.
See Presentation for more detail.
Diagnosis
Laboratory studies include the following:
-
Histopathology - Demonstrates an intraepidermal blister; the earliest changes consist of intercellular edema with loss of intercellular attachments in the basal layer
-
Indirect immunofluorescence (IDIF) - If DIF results are positive; circulating intercellular antibodies are detected with IDIF in 80-90% of patients with pemphigus vulgaris [5]
See Workup for more detail.
Management
The aim of pharmacologic therapy for pemphigus vulgaris is to reduce inflammatory response and autoantibody production. Medications used in the disease’s treatment include the following:
-
Corticosteroids - Discourage the inflammatory process by inhibiting specific cytokine production
-
Immunosuppressants - Should be considered early in the course of disease as steroid-sparing agents
-
Intravenous immune globulin (IVIG)
See Treatment and Medication for more detail.
Pathophysiology
Pemphigus vulgaris is an autoimmune intraepithelial blistering disease that affects the skin and mucous membranes and is mediated by circulating autoantibodies directed against keratinocyte cell surfaces. In 1964, autoantibodies against keratinocyte surfaces were described in patients with pemphigus. Clinical and experimental observations indicated that the circulating autoantibodies are pathogenic. An immunogenetic predisposition has been well established.
Blisters in pemphigus vulgaris are associated with the binding of immunoglobulin G (IgG) autoantibodies to keratinocyte cell surface molecules. These intercellular or pemphigus vulgaris antibodies bind to keratinocyte desmosomes and to desmosome-free areas of the keratinocyte cell membrane. The binding of autoantibodies results in a loss of cell-to-cell adhesion, a process termed acantholysis. The antibody alone is capable of causing blistering without complement or inflammatory cells.
Pemphigus vulgaris antigen
Intercellular adhesion in the epidermis involves several keratinocyte cell surface molecules. Pemphigus antibody binds to the keratinocyte cell surface molecules desmoglein 1 (DSG1) and desmoglein 3 (DSG3). The binding of antibody to DSG may have a direct effect on desmosomal adherence or may trigger a cellular process that results in acantholysis. Antibodies specific for nondesmosomal antigens also have been described in the sera of patients with pemphigus vulgaris; however, the role of these antigens in the pathogenesis of pemphigus vulgaris has not been established.
Antibodies
Patients with the mucocutaneous form of pemphigus vulgaris have pathogenic anti-DSG1 and anti-DSG3 autoantibodies. Patients with the mucosal form of pemphigus vulgaris have only anti-DSG3 autoantibodies. Patients with active disease have circulating and tissue-bound autoantibodies of both IgG1 and IgG4 subclasses.
More than 80% of patients with active disease produce autoantibodies to DSG. Disease activity correlates with antibody titers in most patients. In patients with pemphigus vulgaris, the presence of anti-DSG1 autoantibodies, as determined by enzyme-linked immunosorbent assay (ELISA), is more closely correlated with the course of the disease than the presence of anti-DSG3 autoantibodies. Lack of in-vivo antibody binding (reversion to a negative result on direct immunofluorescence [DIF]) is the best indicator of remission and can help predict a lack of flaring when therapy is tapered.
Complement
Pemphigus antibody fixes components of complement to the surface of epidermal cells. Antibody binding may activate complement with the release of inflammatory mediators and recruitment of activated T cells. T cells are clearly required for the production of the autoantibodies, but their role in the pathogenesis of pemphigus vulgaris remains poorly understood. Interleukin (IL)-2 is the main activator of T lymphocytes, and increased soluble receptors have been detected in patients with active pemphigus vulgaris.
Etiology
Although the cause of pemphigus vulgaris remains unknown, several potentially relevant factors have been identified, as follows.
Genetic factors
Predisposition to pemphigus is linked to genetic factors. [9] Certain major histocompatibility complex (MHC) class II molecules—in particular, alleles of human leukocyte antigen (HLA) DR4 (DRB1*0402) and HLA DRw6 (DQB1*0503)—are common in patients with pemphigus vulgaris. [10, 11, 12, 13]
One study found that the genes FGA (fibrinogen alpha chain), VWF (von Willebrand factor), and ACTG1 (actin gamma 1) were dysregulated in patients with pemphigus vulgaris and suggested that such dysregulation may play a role in the pathogenesis of the disease. [14]
Age
Peak age of onset is between 50 and 60 years. Infants with neonatal pemphigus remit with clearance of maternal autoantibodies. The disease may develop in children or in older persons.
Disease association
Pemphigus occurs in patients with other autoimmune diseases, particularly myasthenia gravis (MG) and thymoma. A study of 110 patients with pemphigus found four patients with autoimmune thyroid disease and three patients with rheumatoid arthritis (RA). [15] In this study, however, autoimmune diseases were no more common in 969 first-degree relatives of patients with pemphigus than in the general population.
Environmental factors
A case-control study suggested that the following may be associated with an increased risk of developing pemphigus vulgaris [16] :
-
Consumption of foods containing thiol groups (eg, leeks and tomatoes)
-
High mental stress levels
-
Consumption of certain supplements (eg, calcium and multivitamins)
-
Use of chemical cleaning products that contain lime
Epidemiology
United States and international statistics
Pemphigus vulgaris is uncommon in the United States, and the exact incidence and prevalence depend on the population studied.
Pemphigus vulgaris has been reported to occur worldwide. The incidence of this condition has been reported to be in the range of 0.5-3.2 cases per 100,000 population. It is higher in patients of Ashkenazi Jewish descent and those of Mediterranean origin. Few familial cases have been reported. As with endemic pemphigus, there is some evidence to suggest clustering near industrial sites. [17]
Age-, sex-, and race-related demographics
Although most cases of pemphigus vulgaris occur between the ages of 50 and 60 years, the range is broad, and disease onset in older individuals and in children has been described. Patients are younger at presentation in India than they are in Western countries. [18]
The male-to-female ratio is approximately equal. In adolescence, girls are more likely to be affected than boys.
Pemphigus vulgaris affects persons of all races and ethnic groups. As noted, it is more prevalent in regions where the Jewish population is predominant [19] and in Mediterranean regions. For example, in Jerusalem, the prevalence of pemphigus vulgaris has been estimated at 1.6 cases per 100,000 population, whereas in Connecticut, the prevalence has been reported as 0.42 cases per 100,000 population. [20]
The incidence in the United Kingdom is 0.68 case per 100,000 persons per year. The incidence of pemphigus vulgaris in Tunisia is estimated at 2.5 cases (women, 3.9; men, 1.2) per million population per year, whereas in France, the incidence is 1.3 cases per million population per year (no significant difference between men and women). [21] In Finland, where few people of Jewish or Mediterranean origin live, the prevalence is low, at 0.76 case per million population. [22]
Prognosis
The severity and natural history of pemphigus vulgaris are variable. Before the advent of steroids, most patients with pemphigus vulgaris died. Treatment with systemic steroids has reduced the mortality dramatically, [23] to approximately 5-15%. [2] If not properly treated, pemphigus vulgaris still is often fatal because of the susceptibility to infection and fluid and electrolyte disturbances.
Mortality in patients with pemphigus vulgaris is three times higher than that in the general population. Most deaths occur during the first few years of disease; if the patient survives 5 years, the prognosis is good. Early disease probably is easier to control than widespread disease, and mortality may be higher if therapy is delayed. Complications secondary to the use of high-dose corticosteroids contribute to mortality as well.
Morbidity and mortality are related to the extent of disease, the maximum dose of prednisolone required to induce remission, and the presence of other diseases. The outlook is worse in older patients and in patients with extensive disease. Prognosis is usually better in childhood than in adulthood.
Pemphigus vulgaris involves mucosa in 50-70% of patients. This may limit oral intake secondary to dysphagia. Blistering and erosions secondary to the rupture of blisters may be painful and may limit the patient's daily activities. Patients with pemphigus vulgaris typically heal without scarring unless the disease is complicated by severe secondary infection.
Reversion of DIF to negative can be useful for predicting sustained remission after withdrawal of medication. Plucked hairs are an alternative to skin biopsy in providing a specimen for immunofluorescence; the pilar sheath epithelium of the anagen hair typically demonstrates immunofluorescence comparable to skin. DIF on plucked hairs may be more acceptable to the patient than serial skin biopsies. [24, 25]
Relapses may occur in more than 50% of patients with pemphigus (including variants other than pemphigus vulgaris). In one study, the following were found to be risk factors for relapse [26] :
-
Extensive body surface area (BSA) involvement
-
High patient body mass index (BMI)
-
High degree of severity at onset, as determined by the Pemphigus Disease Area Index (PDAI)
-
Delayed provision of treatment
-
High titers of anti-DSG1 and anti-DSG3 after treatment
A few rare cases of pemphigus vulgaris transitioning to pemphigus foliaceus have been reported.
Patient Education
Because of the chronic nature of pemphigus vulgaris, it is important for patients to have a good understanding of the disease and to be educated regarding its management.
Patients should be advised to minimize skin trauma because their skin will be more fragile than usual as a result of both the disease itself and the use of topical and systemic steroids to treat it.
They should also be educated regarding their medications. If patients are informed about dosages, adverse effects, and symptoms of toxicity, they will be better able to report any adverse effects to the physician.
Finally, instructions regarding appropriate wound care should be provided.
-
Early, small blister filled with clear fluid arises on healthy skin.
-
Flaccid blister filled with clear fluid arises on healthy skin.
-
An erosion.
-
Erosions and healing areas on the back.
-
Healing areas on the chest and abdomen.
-
Direct immunofluorescence showing intercellular immunoglobulin G throughout the epidermis of a patient with pemphigus vulgaris.
