Background
Keratosis follicularis, also known as Darier disease (DD) or Darier-White disease, is an autosomal dominantly inherited genodermatosis characterized by greasy hyperkeratotic papules in seborrheic regions (see the image below), nail abnormalities, and mucous membrane changes. The disease was first reported independently by Darier and White in 1889. White was first to recognize the genetic nature of keratosis follicularis by noticing that a mother and her daughter were affected.
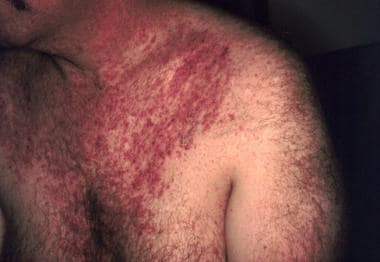 Typical distribution of keratotic papules in seborrheic regions. Image from Susan Mallory, MD, Director of Pediatric Dermatology, Washington University School of Medicine.
Typical distribution of keratotic papules in seborrheic regions. Image from Susan Mallory, MD, Director of Pediatric Dermatology, Washington University School of Medicine.
Several variants of the disease have been described.
Pathophysiology
Mutations in ATP2A2, located on 12q23-24.1, cause keratosis follicularis (Darier disease). [1] ATP2A2 encodes the sarcoplasmic/endoplasmic reticulum Ca2+-ATP isoform 2 protein (SERCA2), which is a calcium pump. This pump maintains a low cytoplasmic Ca2+ level by actively transporting calcium ions from the cytosol into the lumen of the endoplasmic reticulum.
Although more than 250 mutations in ATP2A2 have been identified in patients with Darier disease, [2] attempts at genotype-phenotype correlation have been unsuccessful. Some authors have suggested that recurrent ATP2A2 p.(Pro602Leu) mutation differentiates acrokeratosis verruciformis of Hopf from the allelic condition Darier disease. [3]
Family members with confirmed identical ATP2A2 mutations can exhibit differences in the clinical severity of disease, suggesting that other genes or environmental factors affect the expression of keratosis follicularis. [4, 5] Because the mutation may be inherited in an autosomal dominant fashion or occur sporadically, a negative family history does not exclude the disease. The expressivity of the disease is highly variable, so that a family member may have disease so subtle as to have been overlooked; this further underscores the unreliability of a negative family history.
The mechanisms by which specific ATP2A2 mutations impact the function of the ATP2A2 protein have been investigated via an in-vitro model. [6] Investigators transfected a fibroblast cell line with 51 different mutations seen in Darier disease pedigrees. The resultant transfected cells showed defects in ATP2A2 protein expression (15 mutants), ATP hydrolysis (29 mutants), calcium transport (4 mutants), and calcium binding and kinetics (3 mutants).
In a different study, in which researchers systematically analyzed mutations identical to those found in patients with Darier disease, mutant SERCA2 protein aggregates were found to cause stress to the endoplasmic reticulum, subsequently inducing cell apoptosis. [7] Thus, diverse biochemical mechanisms are responsible for altered protein function.
Although expressivity is variable, penetrance of keratosis follicularis is high, estimated at 95%. Because the disease-causing mutations in ATP2A2 affect functional domains of the gene, the mechanism of autosomal dominant transmission is believed to be haploinsufficiency, in which a single wild-type functioning ATP2A2 is insufficient to prevent disease. No unique phenotype for genetic homozygotes has been reported.
Abnormal keratinocyte-keratinocyte adhesion and aberrant epidermal keratinization are the primary histologic features of keratosis follicularis. Electron microscopy reveals loss of desmosomes (epithelial intercellular junctions formed by membrane and submembrane protein complexes), breakdown of desmosome-keratin intermediate filament attachment, and perinuclear aggregates of keratin intermediate filaments. The mechanism by which decreased activity of the SERCA2 calcium pump leads to these changes remains a subject of investigation. [8] However, a significant correlation exists between the clinical presentation of Darier disease and the intensity of histologic features. [9]
Some studies of keratosis follicularis have suggested that alterations in calcium regulation may affect the synthesis, folding, or trafficking of desmosomal proteins. [10] In particular, studies revealed that Darier disease keratinocytes displayed abnormal trafficking of the desmosomal protein desmoplakin and abnormal expression of cytokeratins 10 and 14. [11, 12] One study showed that SERCA2-controlled Ca²+-dependent keratinocyte adhesion and differentiation are mediated via the sphingolipid pathway. [13]
An alternative hypothesis, based on a canine model of keratosis follicularis, is that in this disease, calcium dysregulation leads to impaired control of cell cycle checkpoints, leading in turn to increased epidermal sensitivity to skin trauma and subsequent keratinocyte apoptosis.
Two particular ATP receptors have been reported to localize abnormally in vivo in patients with keratosis follicularis, and it has been speculated that they play a role in apoptosis as well as abnormal calcium signaling. [12] Subsequently, Darier keratinocytes have been found to display a constitutive endoplasmic reticulum stress response, with immature adherens junctions and desmosomes, which results in decreased intercellular adhesion strength. [14]
Remarkably, an orphan drug, the α-glucosidase inhibitor miglustat, restores mature adherens junctions and desmosomes in Darier keratinocytes and increases adhesion strength. The observation that miglustat can restore proper localization to the plasma membrane of nonmutated proteins retained in the endoplasmic reticulum supports a misfolding mechanism. [14]
Epidemiology
International statistics
Keratosis follicularis (Darier disease) occurs worldwide. Estimates of its prevalence have ranged from 1 case per 30,000 population in Scotland to 1 case per 100,000 population in Denmark.
Age- and sex-related demographics
Keratosis follicularis most commonly manifests from age 6-20 years; however, patients have presented as early as age 4 years and as late as age 70 years. Notably, the first case of congenital Darier disease was diagnosed by means of biopsy in a child with a significant positive family history for the disease, in which at least the three preceding generations of family members were affected. [15]
Males and females are equally affected by keratosis follicularis.
Prognosis
Patients with keratosis follicularis (Darier disease) experience pruritus and sometimes pain in the affected skin areas. Psychosocial consequences from the appearance and odor of the lesions also constitute the major morbidity of Darier disease.
A serious complication associated with keratosis follicularis is increased susceptibility to cutaneous bacterial and viral infections, in particular herpes simplex virus (HSV), human papillomavirus (HPV), [16] and poxvirus infections. Initial misdiagnosis of Darier disease may result in undertreatment of such infections, potentially leading to fatal outcomes. [17, 18] Overall, however, patients with keratosis follicularis have a life expectancy similar to that of the general population.
Neuropsychiatric abnormalities (eg, epilepsy, mental impairment, schizophrenia, [19] and mood disorders) have been associated with keratosis follicularis. Several national studies have suggested that genetic variability within the ATP2A2 gene that causes Darier disease also confers susceptibility to a number of neuropsychiatric disorders, [20] including bipolar disorder, [21] intellectual disability, and subclinical impairments in cognitive ability. [22]
Patient Education
Darier disease is prone to exacerbations induced by infection, heat, and friction. Adoption of measures to avoid precipitating factors is recommended where possible.
Genetic counseling is recommended so that patients are aware of the risk to potential offspring.
-
Typical distribution of keratotic papules in seborrheic regions. Image from Susan Mallory, MD, Director of Pediatric Dermatology, Washington University School of Medicine.
-
Longitudinal ridges, red and white lines, and V-shaped nicks. Image from Susan Mallory, MD, Director of Pediatric Dermatology, Washington University School of Medicine.
-
Acantholysis and dyskeratosis (abnormal keratinization) are two main features of Darier disease. Loss of epidermal adhesion with acantholysis frequently results in formation of suprabasal clefts (lacunae).
Tables

What would you like to print?
- Tailor Actinic Keratosis Care for the Immunocompromised
- DIY: Seborrheic Keratosis Eradication
- Transplant Recipients Face High Risk for Actinic Keratosis
- Interim Clinical Treatment Considerations for Severe Manifestations of Mpox
- Use of JYNNEOS (Smallpox and Monkeypox Vaccine, Live, Nonreplicating) for Preexposure Vaccination of Persons at Risk for Occupational Exposure to Orthopoxviruses
-
 Despite New Ichthyosis Treatment Recommendations, 'Many Questions Still Exist'
Despite New Ichthyosis Treatment Recommendations, 'Many Questions Still Exist'

