Practice Essentials
Autosomal dominant hyper-IgE syndrome (AD-HIES), formerly known as Job syndrome, is a rare syndrome that includes the triad of eosinophilia, eczema, and recurrent skin and pulmonary infections. Mutations in the STAT3 gene cause most cases of AD-HIES. Elevated IgE level is a cardinal feature of the syndrome. [1, 2]
Signs and symptoms
Symptoms often become apparent early during infancy or childhood. AD-HIES is characterized by repeated bacterial infections of the skin (eczema) and lungs (pneumonia), skeletal abnormalities, and characteristic facial features. Skeletal abnormalities include scoliosis, joint hyperextensibility, osteoporosis, and repeated fractures of the long bones of the arms and legs and the ribs. Characteristic facial features include a broad nasal bridge, deep-set eyes, prominent forehead, and irregularly proportioned cheeks and jaws, and generalized hardening (coarsening) of the skin.
Diagnosis
Blood tests demonstrate elevated levels of IgE in the blood [3] and elevated levels of eosinophils (eosinophilia). Chest x-ray, chest CT, or both are necessary to determine the extent of pulmonary parenchymal involvement.
Management
Care of patients with AD-HIES is primarily directed at treating and preventing recurrent skin and sinopulmonary infections. Currently, no clear recommendations exist for routine medical therapy of the vascular, musculoskeletal, and connective tissue abnormalities associated with the disorder.
Background
Autosomal dominant hyperimmunoglobulin E (IgE) syndrome (AD-HIES) was first described as Job syndrome in 1966 [1, 2] and included the triad of eosinophilia, eczema, and recurrent skin and pulmonary infections. Elevated IgE level was recognized as a cardinal feature of the syndrome in 1972, and the name HIES was subsequently proposed. [3] The phenotype was later expanded to include many connective tissue and skeletal abnormalities. A scoring system that weighted both the immunologic and somatic features of the syndrome was designed to aid in the clinical diagnosis of these patients (see the table below). [4]
Table. Scoring System for Job Syndrome (Open Table in a new window)
|
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
10 |
Clinical Findings |
|
|
|
|
|
|
|
|
|
|
Highest IgE (IU/mL) |
< 200 |
200-500 |
|
|
501-1000 |
|
|
|
1001-2000 |
>2000 |
Total # skin abscesses/boils |
None |
|
1-2 |
|
3-4 |
|
|
|
>4 |
|
Total # pneumonias |
None |
|
1 |
|
2 |
|
3 |
|
>3 |
|
Parenchymal lung abnormalities |
None |
|
|
|
|
|
Bronchiectasis |
|
Pneumatocele |
|
Other serious infection |
None |
|
|
|
Present |
|
|
|
|
|
Fatal infection |
None |
|
|
|
Present |
|
|
|
|
|
Highest eosinophils/uL |
< 700 |
|
|
701-800 |
|
|
>800 |
|
|
|
Newborn rash |
None |
|
|
|
Present |
|
|
|
|
|
Eczema (worst stage) |
None |
Mild |
Moderate |
|
Severe |
|
|
|
|
|
Sinusitis/otitis (# in worst year) |
1-2 |
3 |
4-6 |
|
>6 |
|
|
|
|
|
Candidiasis |
None |
Oral, vaginal |
Fingernail |
|
Systemic |
|
|
|
|
|
Retained primary teeth |
None |
1 |
2 |
|
3 |
|
|
|
>3 |
|
Scoliosis (max. curvature) |
< 10 |
|
10-14 |
|
15-20 |
|
|
|
>20 |
|
Minimal trauma fractures |
None |
|
|
|
1-2 |
|
|
|
>2 |
|
Hyperextensibility |
None |
|
|
|
Present |
|
|
|
|
|
Characteristic face |
None |
|
Mild |
|
|
Present |
|
|
|
|
Increased interalar distance |
< 1 SD |
1-2 SD |
|
>2 SD |
|
|
|
|
|
|
High palate |
None |
|
Present |
|
|
|
|
|
|
|
Congenital anomaly |
None |
|
|
|
|
Present |
|
|
|
|
Lymphoma |
None |
|
|
|
Present |
|
|
|
|
|
More recently, vascular abnormalities, including coronary aneurysm without atherosclerosis, and brain MRI abnormalities, including focal hyperintensities and Chiari I malformations, have been described. In 2007, autosomal dominant mutations in signal transducer and activator of transcription-3 (STAT3) gene were identified as the molecular cause of this disease. [5, 6, 7] See image below.
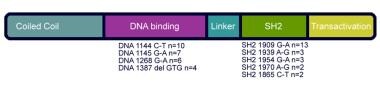 STAT3 gene is diagrammed with depiction of hotspots (areas where higher numbers of patients were noted to have mutations).
STAT3 gene is diagrammed with depiction of hotspots (areas where higher numbers of patients were noted to have mutations).
Pathophysiology
Mutations in the STAT3 gene have been identified in almost all cases of clinically verified autosomal dominant hyper-IgE syndrome (AD-HIES). [8, 9] STAT3 is one of 7 human STAT (signal transducer and activator of transcription) proteins, which are critical second messengers for many cytokine, hormone, and growth factor receptors. In general, Janus family tyrosine kinases (JAKs) bind to the intracellular components of cytokine receptors, and are, in turn, bound by STATs upon cytokine signaling. When cytokines bind to their cognate receptor, JAKs phosphorylate the cytokine receptor and subsequently the STATs, which then dissociate from the JAK-receptor complex. Phosphorylated STATs dimerize within the cytosol via their phosphotyrosines and Src-homology 2 (SH2) domains. Dimerized STATs then translocate to the nucleus, where they bind DNA in the promoter sequences of target genes to activate transcription. This activity is typically stopped by STAT dephosphorylation. STAT3 is located on human chromosome17q21.
STAT3 mutations have been found in persons of Asian, African, Caucasian, and Hispanic descent and correlate well with HIES disease. The mutations are predominantly missense and in-frame deletions that lead to production of proteins with dominant negative activity. No null alleles have been identified to date, indicating that haploinsufficiency is not a mechanism for disease causation. Most AD-HIES STAT3 mutations are located in the SH2 and DNA binding regions. Dominant negative mutants antagonize the wild type protein, leading to less than 50% STAT3 activity. This is consistent with the mouse data, which show that heterozygous STAT3 deficiency is not associated with disease, while STAT3 null animals die during embryogenesis. Therefore, HIES patients have less than 50% STAT3 activity, which is enough to sustain life and many aspects of development but inadequate for normal immune function or normal continued tissue remodeling.
AD-HIES is a disease of both too little and too much inflammatory response, which is in part explained by the important role of STAT3 in the induction and signalling of key cytokines, including interleukins IL-6, IL-10, IL-17, IL-22, and IL-23. IL-6 induces acute phase proteins by hepatocytes, neutrophil production in cooperation with colony-stimulating growth factors, stimulation of differentiated B cell growth, stimulation of proinflammatory cytokine production (particularly IL-17), inhibition of the action and production of regulatory T cells, and upregulation of the chemokine MCP-1 (also known as CCL2).
Due to impaired IL-6 and IL-23 signalling through STAT3, the crucial T cell transcription factor retinoid-related orphan receptor gamma (ROR-gt) is diminished, impairing IL-17 expression and Th17 differentiation. [10] Th17 cells are T helper cells that produce IL-17, a cytokine important in neutrophil recruitment and activation and defense against fungal and extracellular bacteria. Th17 cells also produce IL-22, which stimulates epithelial production of b-defensins, small peptides crucial for killing bacteria and fungi. These cytokines, which are inadequately upregulated in HIES, may explain some of the susceptibility to infections of the skin and lung epithelium.
Overexuberant inflammation in AD-HIES is suggested by the frequent development of pneumatoceles following pneumonias, despite adequate therapy of pathogens. Transcriptional array studies and cytokine production studies both show increased proinflammatory cytokines such as tumor necrosis factor alpha (TNFa), interferon gamma (IFNg), and IL-12 in AD-HIES cells compared to normals. Abnormally low IL-10, a cytokine critical for dampening the inflammatory response, may also contribute to the exuberant inflammatory response in HIES. IL-10 signaling is mediated via STAT3. IL-10 has negative feedback regulation of macrophages, which express costimulators that enhance T cell activation and secrete cytokines such as IL-12 and IL-23 to enhance cell-mediated immunity. Therefore, impaired IL-10 production and signaling may be associated with increased proinflammatory cytokines. It may even contribute to the increased IgE and eosinophilia in AD-HIES due to the lack of normal IL-4 suppression.
Epidemiology
Frequency
Autosomal dominant hyper-IgE syndrome (AD-HIES) is a rare disease. The exact incidence is unknown. It has no known associations with race, ethnicity, or gender.
Mortality/Morbidity
AD-HIES is associated with a contracted lifespan, with deaths occurring predominantly in adulthood due to the consequences of chronic infections. In a recent review of a large cohort of patients, the average living patient age was 27 years, but ages ranged from 3 to 58 years. Infection-related deaths occurred at an average age of 29 years.
Death in AD-HIES patients is most commonly due to chronic pulmonary infections in the setting of pneumatoceles or bronchiectasis or both. Molds, such as Aspergillus and Scedosporium species, may invade the pulmonary blood vessels, leading to metastatic spread or fatal hemoptysis. These infections are secondary opportunists in regions of destroyed lung caused by the inappropriate healing of previous infections. [11]
AD-HIES patients are at increased risk for Hodgkin and non-Hodgkin lymphoma (relative risk 259), although the number of cases reported is small. [12]
Race
STAT3 mutations have been detected in persons of Caucasian, African, Asian, and Hispanic descent. Although most documented cases are in white patients, this is likely due to referral bias of the population studied.
Sex
No gender-based differences have been described with regard to disease severity or incidence.
Age
In a recent chart review, the average age of diagnosis was 11.5 years. However, the diagnosis can be made in infancy or adulthood. Proband survival has improved over the years along with improved management, allowing patients to have their own children. Given the autosomal dominant nature of HIES, these offspring of known patients receive early diagnosis and therapy.
-
STAT3 gene is diagrammed with depiction of hotspots (areas where higher numbers of patients were noted to have mutations).
-
HIES scoring sheet. A score >40 is considered likely HIES, 20-40 indeterminate, < 20 unlikely HIES.
-
Recurrent pneumonias, particularly those due to S aureus, may lead to pneumatocele formation. Pneumatoceles such as those demonstrated in this CT image may then allow fungal superinfection.
-
Brain hyperintensities have been noted to be present with increased frequency in patients of all ages with Job syndrome.
Tables
|
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
10 |
Clinical Findings |
|
|
|
|
|
|
|
|
|
|
Highest IgE (IU/mL) |
< 200 |
200-500 |
|
|
501-1000 |
|
|
|
1001-2000 |
>2000 |
Total # skin abscesses/boils |
None |
|
1-2 |
|
3-4 |
|
|
|
>4 |
|
Total # pneumonias |
None |
|
1 |
|
2 |
|
3 |
|
>3 |
|
Parenchymal lung abnormalities |
None |
|
|
|
|
|
Bronchiectasis |
|
Pneumatocele |
|
Other serious infection |
None |
|
|
|
Present |
|
|
|
|
|
Fatal infection |
None |
|
|
|
Present |
|
|
|
|
|
Highest eosinophils/uL |
< 700 |
|
|
701-800 |
|
|
>800 |
|
|
|
Newborn rash |
None |
|
|
|
Present |
|
|
|
|
|
Eczema (worst stage) |
None |
Mild |
Moderate |
|
Severe |
|
|
|
|
|
Sinusitis/otitis (# in worst year) |
1-2 |
3 |
4-6 |
|
>6 |
|
|
|
|
|
Candidiasis |
None |
Oral, vaginal |
Fingernail |
|
Systemic |
|
|
|
|
|
Retained primary teeth |
None |
1 |
2 |
|
3 |
|
|
|
>3 |
|
Scoliosis (max. curvature) |
< 10 |
|
10-14 |
|
15-20 |
|
|
|
>20 |
|
Minimal trauma fractures |
None |
|
|
|
1-2 |
|
|
|
>2 |
|
Hyperextensibility |
None |
|
|
|
Present |
|
|
|
|
|
Characteristic face |
None |
|
Mild |
|
|
Present |
|
|
|
|
Increased interalar distance |
< 1 SD |
1-2 SD |
|
>2 SD |
|
|
|
|
|
|
High palate |
None |
|
Present |
|
|
|
|
|
|
|
Congenital anomaly |
None |
|
|
|
|
Present |
|
|
|
|
Lymphoma |
None |
|
|
|
Present |
|
|
|
|
|

What would you like to print?
- Nemolizumab Effective in Atopic Dermatitis
- Allergic Contact Dermatitis: New Culprits
- Inflammation Now the Key Target for Seborrheic Dermatitis
-
 What's New in Psoriasis? Dermasphere Podcasters Weigh In
What's New in Psoriasis? Dermasphere Podcasters Weigh In
-
American Academy of Dermatology (AAD) 2025 Annual Meeting
-
More Than Hives: Discussing Chronic Spontaneous Urticaria







