Practice Essentials
Dermatomyositis is an idiopathic inflammatory myopathy with characteristic cutaneous findings that occur in children and adults (see the image below). This systemic disorder most frequently affects the skin and muscles but may also affect the joints; the esophagus; the lungs; and, less commonly, the heart. [1, 2] Dystrophic calcinosis may complicate dermatomyositis and is most often observed in children and adolescents.
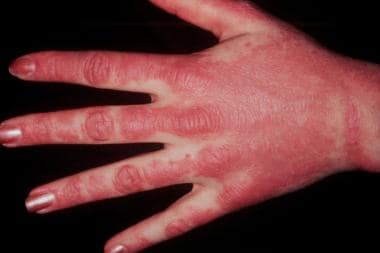 These lesions on dorsal hands demonstrate photodistribution of dermatomyositis. Note sparing of interdigital web spaces.
These lesions on dorsal hands demonstrate photodistribution of dermatomyositis. Note sparing of interdigital web spaces.
Signs and symptoms
Persons with dermatomyositis often present with skin disease as one of the initial manifestations, and it may be the sole manifestation at onset in perhaps as many as 40% of individuals with this condition. Cutaneous involvement may manifest as follows:
-
Eruption predominantly on photo-exposed surfaces
-
Pruritus of skin lesions, sometimes intense enough to disturb sleep
-
Erythema of the mid-face
-
Eruption along the eyelid margins, with or without periorbital edema
-
Eruption on the dorsal hands, particularly over the knuckles
-
Changes in the nailfolds of the fingers
-
Eruption of the upper outer thighs
-
Scaly scalp or diffuse hair loss [3]
Muscle disease may occur concurrently, may precede the skin disease, or may follow the skin disease by weeks to years. Muscle involvement manifests as the following:
-
Proximal muscle weakness
-
Muscle fatigue/weakness when climbing stairs, walking, rising from a seated position, combing hair, or reaching for items above shoulders
-
Muscle tenderness: May occur, but not a typical feature of dermatomyositis
Systemic manifestations that may occur include the following:
-
General systemic disturbances, fever, arthralgia, malaise, weight loss, Raynaud phenomenon
-
Dysphagia due to esophageal skeletal muscle involvement
-
Gastroesophageal reflux due to esophageal smooth muscle involvement
-
Dysphonia
-
Atrioventricular defects, tachyarrhythmias, dilated cardiomyopathies
-
Gastrointestinal ulcers and infections, more common in children
-
Pulmonary involvement due to weakness of thoracic muscles, interstitial lung disease
-
Subcutaneous calcification, [4] which may result in contracture of joints; more common in children
-
Children may also develop a tiptoe gait secondary to flexion contracture of the ankles in early childhood
See Presentation for more detail.
Diagnosis
Examination for cutaneous dermatomyositis may reveal the following findings:
-
Characteristic, possibly pathognomonic cutaneous features: Heliotrope, Gottron papules
-
Characteristic but not pathognomonic features: Malar erythema, violaceous erythema or poikiloderma in a photosensitive distribution, violaceous erythema on the extensor surfaces, and periungual and cuticular changes
-
Violaceous erythema or poikiloderma involving the anterior chest is referred to as the “V-neck sign” whereas involvement of the upper back and shoulders is referred to as the “shawl sign”
-
Rare cutaneous manifestations include vesiculobullous erosive lesions and an exfoliative erythroderma, which may be more common in patients with an associated malignancy than in those without a malignancy; biopsy samples of these manifestations reveal an interface dermatitis similar to that seen in biopsy samples of heliotrope rash, Gottron papules, poikiloderma, or scalp lesions
Examination for muscle disease in dermatomyositis may demonstrate the following:
-
Quadriparesis involving proximal musculature
-
Difficulty rising from a seated or supine position without support
-
Extensor muscles often more affected than the flexor muscles
-
Neck flexor muscle weakness
-
Distal strength, sensation, and tendon reflexes maintained (unless the patient has severely weak and atrophic muscle)
Testing
Laboratory and other studies that may be helpful include the following:
-
Muscle enzyme levels (eg, creatine kinase, aldolase, aspartate aminotransferase, lactate dehydrogenase)
-
Myositis-specific antibodies
-
Antinuclear antibody levels
-
Pulmonary function studies with diffusion capacity
-
Electrocardiography
-
Esophageal manometry
-
Colonoscopy to screen for underlying malignancy
-
Papanicolaou smear in women for malignancy screening
-
CA-125 and CA-19-9 for malignancy screening
Imaging studies
The following imaging studies may be used in the evaluation of dermatomyositis:
-
MRI or ultrasonography of the muscles
-
Chest radiography
-
Barium swallow
-
Electromyography
-
Imaging to screen for underlying malignancy, including CT scanning of the chest, abdomen, and pelvis, as well as transvaginal ultrasound and mammography for women
Procedures
The following procedures may be helpful in the evaluation of dermatomyositis:
-
Skin biopsy
-
Muscle biopsy (open or via a needle): Findings can be diagnostic (perivascular and interfascicular inflammatory infiltrates with adjoining groups of muscle fiber degeneration/regeneration)
See Workup for more detail.
Management
Therapy for the muscle component of dermatomyositis involves the use of corticosteroids, typically with an immunosuppressive agent. Therapy for the skin disease includes the following, among other options:
-
Sun avoidance
-
Sunscreens and photoprotective clothing
-
Topical corticosteroids
-
Antimalarial agents
-
Methotrexate
-
Mycophenolate mofetil
-
Immune globulins
Pharmacotherapy
Medications used in the management of dermatomyositis include the following:
-
Corticosteroids (eg, prednisone): Prednisone is a first-line therapy for muscle involvement in dermatomyositis
-
Immunosuppressive agents (eg, methotrexate, mycophenolate mofetil, azathioprine, rituximab, sirolimus)
-
Immune globulins (eg, intravenous or subcutaneous immunoglobulin)
-
Antimalarial agents (eg, hydroxychloroquine, chloroquine)
Rituximab may be useful in the treatment of muscle disease in dermatomyositis, and has had mixed results in treatment of skin disease. [11, 12, 13] In addition to the medications listed above, diltiazem, colchicine, alendronate, and warfarin are among the medications that have shown potential benefit in treating calcinosis. Surgical excision of focal, tender calcinotic lesions is also considered a therapeutic option.
Nonpharmacotherapy
General therapeutic measures may include the following:
-
Physical therapy and rehabilitative measures
-
Sun avoidance
-
Sun-protection (eg, broad-spectrum sunscreens, sun protective clothing)
-
Elevation of head of bed
-
Avoidance of eating before bedtime
Surgery
Surgical care is usually unnecessary in the management of dermatomyositis. However, some patients may benefit from surgical removal of localized areas of calcinosis, particularly those that are painful.
See Treatment and Medication for more detail.
The prognosis of dermatomyositis depends on the severity of the myopathy, the presence of malignancy, and/or the presence of esophageal and/or cardiopulmonary involvement. Residual weakness is common, even in patients who fully recover.
For discussion of dermatomyositis in pediatric patients, see Juvenile Dermatomyositis. For patient education information, see Dermatomyositis and The Myositis Association Web site.
Background
In 1975, Bohan and Peter first suggested a set of five criteria to aid in the diagnosis and classification of dermatomyositis. [14, 15] Four of the five criteria are related to the muscle disease, as follows:
-
Progressive proximal symmetrical weakness
-
Elevated levels of muscle enzymes
-
An abnormal finding on electromyography
-
An abnormal finding on muscle biopsy
The fifth criterion is compatible cutaneous disease.
In addition to dermatomyositis, Bohan and Peter suggested the following four subsets of myositis [15] :
-
Polymyositis
-
Myositis with malignancy
-
Childhood dermatomyositis/polymyositis
-
Myositis overlapping with another collagen-vascular disorder
In a subsequent publication, Bohan et al noted that cutaneous disease may precede the development of the myopathy in patients with dermatomyositis. [14] In addition, the existence of another subset of patients with dermatomyositis that affects only the skin (ie, amyopathic dermatomyositis [ADM], or dermatomyositis sine myositis) has been recognized. Finally, another subset of patients with dermatomyositis are those with controlled myopathy who continue to have severe and sometimes debilitating skin disease (ie, postmyopathic dermatomyositis).
ADM is diagnosed in patients with typical cutaneous disease who show no evidence of muscle weakness and in whom serum muscle enzyme levels are repeatedly normal over a 2-year period in the absence of the use of disease-modifying therapies such as corticosteroids, immunosuppressive agents, or both for 2 months or longer.
When studied, some ADM patients may have abnormal findings on ultrasonography, electromyography, magnetic resonance imaging (MRI), magnetic resonance spectroscopy, or muscle biopsy. These patients are better classified as having hypomyopathic dermatomyositis. ADM or hypomyopathic dermatomyositis may also be related to an underlying malignancy.
The term clinically amyopathic dermatomyositis (CADM) is often used to encompass patients with both amyopathic and hypomyopathic dermatomyositis. [16] CADM is estimated to account for about 20% of patients with dermatomyositis, [17] and one large review suggests that CADM is associated with malignancy and lung disease as frequently as classic dermatomyositis. [18] In addition, some patients with CADM develop severe pulmonary disease, particularly persons from Asian countries. [19]
Patients exist in whom myositis resolves after therapy but skin disease remains an active and important feature of the disorder. These patients are not classified as having ADM, even though by this point the skin lesions are the major and often only manifestation of the disease. Germani and colleagues have suggested the term postmyopathic dermatomyositis for these patients. [20]
Pathophysiology
Dermatomyositis is considered to be the result of a humoral attack against the muscle capillaries and small arterioles (endothelium of the endomysial blood vessels). Since 1966, there has been evidence supporting an ongoing microangiopathy. [21]
Like other idiopathic inflammatory myopathies, dermatomyositis is characterized by the presence of autoantibodies that are conventionally divided into myositis-specific autoantibodies (MSAs) and myositis-associated autoantibodies (MAAs). Individual patients rarely generate more than one MSA simultaneously. MSAs found in dermatomyositis include anti–Mi-2, anti-MDA5, anti–TIF1γ, anti-NXP2, and anti-SAE. [22]
The disease starts when putative antibodies or other factors activate C3, forming C3b and C4b fragments that lead to formation of C3bNEO and membrane attack complex (MAC), which are deposited in the endomysial vasculature. Complement C5b-9 MAC is deposited and is needed in preparing the cell for destruction in antibody-mediated disease. B cells and CD4 (helper) cells are also present in abundance in the inflammatory reaction associated with the blood vessels.
As the disease progresses, the capillaries are destroyed, and the muscles undergo microinfarction. Perifascicular atrophy occurs in the beginning; however, as the disease advances, necrotic and degenerative fibers are present throughout the muscle.
The pathogenesis of the cutaneous component of dermatomyositis is poorly understood, but is thought to be similar to that of muscle involvement.
Studies on the pathogenesis of the muscle component have been controversial. Some suggest that the myopathy in dermatomyositis is pathogenetically different from that in polymyositis. The former is probably caused by complement-mediated (terminal attack complex) vascular inflammation, the latter by the direct cytotoxic effect of CD8+ lymphocytes on muscle. However, other cytokine studies suggest that some of the inflammatory processes may be similar. One report has linked tumor necrosis factor (TNF) abnormalities with dermatomyositis. [23]
Etiology
The cause of dermatomyositis is unknown. However, genetic, immunologic, infectious, and environmental factors have been implicated.
A genetic component may predispose to dermatomyositis. Dermatomyositis rarely occurs in multiple family members, but a link to certain human leukocyte antigen (HLA) types (eg, DR3, DR5, DR7) may exist.
Polymorphisms of tumor necrosis factor (TNF) may be involved; specifically, the presence of the -308A allele is linked to photosensitivity in adults and calcinosis in children. [23, 24, 25] A meta-analysis demonstrated that the TNF-α-308A/G polymorphism might contribute to dermatomyositis susceptibility, especially in a European population. [26]
Immunologic abnormalities are common in patients with dermatomyositis. Patients frequently have circulating autoantibodies. Abnormal T-cell activity may be involved in the pathogenesis of both the skin disease and the muscle disease. In addition, family members may manifest other diseases associated with autoimmunity.
Antinuclear antibodies (ANAs) and antibodies to cytoplasmic antigens (ie, antitransfer RNA synthetases) may be present. Although their presence may help to define subtypes of dermatomyositis and polymyositis, their role in pathogenesis is uncertain.
Infectious agents have been suggested as possible triggers of dermatomyositis. These include the following:
-
Viruses (eg, coxsackievirus, parvovirus, echovirus, human T-cell lymphotropic virus type 1 [HTLV-1], HIV)
-
Toxoplasma species
-
Borrelia species
New cases of dermatomyositis have been reported following COVID-19 infection or vaccination. [27, 28]
Cases of drug-induced dermatomyositis have been reported. Dermatomyositis-like skin changes have been reported with hydroxyurea in patients with chronic myelogenous leukemia or essential thrombocytosis. [29, 30] Other agents that may trigger the disease include the following:
-
Statins
-
Penicillamine
-
Tumor necrosis factor inhibitors [31]
-
Programmed cell death inhibitors [32]
-
Interferon
-
Cyclophosphamide
-
Bacillus Calmette-Guérin (BCG) vaccine
-
Quinidine
-
Phenylbutazone (no longer approved or marketed for human use in the United States)
Dermatomyositis may be initiated or exacerbated by silicone breast implants or collagen injections, but the evidence for this is anecdotal and has not been verified in case-control studies. One report identified a distinct immunogenetic profile in women who developed inflammatory myopathy after receiving silicone implants. [33]
Epidemiology
The estimated incidence of dermatomyositis is 9.63 cases per million population. The estimated incidence of AMD is 2.08 cases per million. [17]
Dermatomyositis can occur in people of any age. Two peak ages of onset exist: in adults, the peak age of onset is approximately 50 years, whereas in children, the peak age is approximately 5-10 years. Dermatomyositis and polymyositis are twice as common in women as in men. Neither condition shows any racial predilection.
Prognosis
Dermatomyositis may spontaneously remit in as many as 20% of affected patients. About 5% of patients have a fulminant progressive course with eventual death. However, patients who survive the disease may experience residual weakness and disability. Children with severe dermatomyositis may develop contractures. Therefore, many patients require long-term therapy.
Risk factors for a poorer prognosis in patients with dermatomyositis include the following:
-
An associated malignancy
-
Cardiac, pulmonary, or esophageal involvement
-
Older age (ie, > 60 years)
Dermatomyositis may cause death because of muscle weakness or cardiopulmonary involvement. [34] Patients with an associated cancer may die of the malignancy.
The association between malignancy and dermatomyositis has long been recognized. An estimated 25% of patients with dermatomyositis have or will develop an associated malignancy, and the risk appears to remain elevated for 3-5 years. [35, 36, 37] Strong data from Scandinavia, Australia, North America, and Asia continue to confirm this association with malignancy, and existing data supports that risk of malignancy is similar in patients with little or no evidence of muscle involvement (clinically amyopathic dermatomyositis [CADM]) and in those with classic dermatomyositis. [18, 20, 36, 37, 38, 39, 40, 41]
The presence of specific circulating autoantibodies has been linked with risk of malignancy in patients with dermatomyositis. The strongest reported correlation is with circulating anti–transcriptional intermediary factor 1 (TIF1), anti-nuclear matrix protein 2 (NXP2), and anti–SUMO-1 activating enzyme (SAE) autoantibodies. [42]
Ovarian cancer is clearly over-represented in patients with dermatomyositis; however, any malignancy may occur. Reported maligancies include lung, colon, prostate, breast, pancreatic, cervical, and hematologic malignancies. [43] Predilection for certain types of malignancy may be more common in specific populations. For example, nasopharyngeal carcinoma appears to be over-represented in certain Asian populations. [44, 45, 46]
In an approximately 10-year retrospective study from southern China, 60 of 246 dermatomyositis patients developed malignancies. The risk of malignancy was highest in the first year after diagnosis of dermatomyositis, and nasopharyngeal carcinoma and ovarian carcinoma were the most common malignancies. Male gender, dysphagia and elevated erythrocyte sedimentation rate were risk factors for malignancy, whereas the presence of interstitial lung disease appeared to reduce the risk of malignancy. [47] Older age appears to be the strongest predictor of malignancy in patients with dermatomyositis.
Calcinosis may also complicate dermatomyositis. It is rare in adults but is more common in children and has been linked to delay in diagnosis and to less-aggressive therapy. [48] Contractures can occur if the patient is immobile.
Population-based studies from British Columbia concluded that patients with dermatomyositis (or polymyositis) are at increased risk for venous thromboembolism (deep venous thrombosis or pulmonary embolism) and myocardial infarction, especially in the first year after diagnosis. [49, 50] However, dermatomyositis was not associated with an increased risk of ischemic stroke. [50]
A study from Taiwan reported that the risk of osteoporosis in persons with dermatomyositis (or polymyositis) was 2.99 times higher than in those without these disorders. This risk was independent of treatment with corticosteroids and immunosuppressant drugs. [51]
In a study of patients with dermatomyositis with cutaneous involvement, 28 of 74 achieved clinical remission of the skin disease during a 3-year follow-up period. Clinical remission of skin disease was more likely to occur in older patients (odds ratio [OR], 1.07; 95% CI, 1.02-1.12; P = 0.01), those with a dermatomyositis-associated malignancy (OR, 14.46; 95% CI, 2.18-96.07; P =0 .01), and those treated with mycophenolate mofetil (OR, 6.00; 95% CI, 1.66-21.78; P = 0.01). Patients with antimelanoma differentiation–associated protein 5 antibodies had a significantly lower probability of achieving clinical remission. [52]
African Americans and patients in lower socioeconomic groups are more likely to experience a delay in diagnosis. The prognosis in children with dermatomyositis is worse in those in whom diagnosis is delayed.
Overall, data suggest that the mortality rate in persons with dermatomyositis is higher than that in the general population. Population-based data from Sweden demonstrate that mortality in idiopathic inflammatory myopathies is increased within 1 year of diagnosis and plateaus around 10 years after diagnosis, with the increase in mortality largely attributed to malignancy, as well as to diseases of the respiratory and vascular systems. [53]
-
Heliotrope flower, for which characteristic manifestation of dermatomyositis is named.
-
Heliotrope rash in a woman with dermatomyositis.
-
Gottron papules and nailfold telangiectasia are present in this patient with dermatomyositis.
-
These lesions on dorsal hands demonstrate photodistribution of dermatomyositis. Note sparing of interdigital web spaces.
-
Diffuse alopecia with scaly scalp dermatosis is common in patients with dermatomyositis.
-
Dermatomyositis is often associated with a poikiloderma in a photodistribution.
-
Histopathology of dermatomyositis is interface dermatitis.
-
Calcinosis caused by dermatomyositis in childhood can be observed in patient who had active dermatomyositis 15 years before time of this photograph.
-
Histopathology of dermatomyositis showing inflammatory myopathic changes with a predominantly perivascular chronic inflammatory infiltrate.
-
Calcifying panniculitis in patient with dermatomyositis.
-
Ulceration over dorsal and lateral fingers in patient with dermatomyositis.
-
Hematoxylin and eosin paraffin section shows polymyositis. Longitudinal section shows dense, chronic, endomysial inflammatory infiltrate. Image courtesy of Roberta J Seidman, MD.
-
Hematoxylin and eosin frozen section shows polymyositis. Endomysial chronic inflammation is present among intact myofibers that are remarkable only for increased variability of fiber size. Image courtesy of Roberta J Seidman, MD.
-
Hematoxylin and eosin paraffin section shows polymyositis. Patient had dense endomysial inflammation that contains abundance of plasma cells, which can be observed in patients with chronic polymyositis. Two necrotic myofibers, characterized by dense eosinophilic staining, are observed. Focal fatty infiltration of muscle is present in lower left quadrant of photomicrograph. Image courtesy of Roberta J Seidman, MD.
-
Hematoxylin and eosin paraffin section shows polymyositis. Photomicrograph illustrates attack on nonnecrotic myofiber by autoaggressive T lymphocytes. On left, central myofiber is intact. On right, it is obliterated by segmental inflammatory attack. If immunohistochemistry were performed, expected findings would include admixture of CD8 T lymphocytes and macrophages in inflammatory process. Image courtesy of Roberta J Seidman, MD.
-
Hematoxylin and eosin paraffin shows dermatomyositis. In dermatomyositis, inflammation is characteristically perivascular and perimysial. Vessel oriented approximately vertically in center has mild perivascular chronic inflammatory infiltrate. Endothelium is plump; wall is not necrotic. A few lymphocytes in wall of vessel are probably in transit from lumen to external aspect of vessel. Some observers may interpret this finding as vasculitis, but it is certainly neither necrotizing vasculitis nor arteritis. Image courtesy of Roberta J. Seidman, MD.
-
Hematoxylin and eosin frozen section shows perifascicular atrophy in dermatomyositis. Fascicles in this sample show atrophy, predominantly at periphery, along connective-tissue border. Ischemia is considered to cause perifascicular atrophy. This finding is characteristic of dermatomyositis, mostly associated with juvenile form but also observed in adult form. Image courtesy of Roberta J Seidman, MD.
-
Immunofluorescence for membrane attack complex of complement (MAC) in dermatomyositis. Bright ring of yellow-green fluorescence at center represents MAC in wall of microvessel. Finding was not present after treatment with steroids. Image courtesy of Roberta J Seidman, MD.
-
A 47-year-old woman presented with a pruritic, diffuse rash across her upper hands and face that is worsened with sun exposure. ANA testing by outside providers was negative. Her rash was not responsive to topical steroids, and improved with oral prednisone but recurred with tapers beyond 15 mg daily. Diagnosis was dermatomyositis sine myositis. Image courtesy of Jason Kolfenbach, MD, and Kevin Deane, MD, Division of Rheumatology, University of Colorado Denver School of Medicine.
Tables
Autoantibody |
Clinical Associations |
Anti–Mi-2 |
Typical dermatomyositis rashes |
Anti–MDA5 |
Particular skin signs Clinically amyopathic dermatomyositis Rapidly progressive interstitial lung disease |
Anti–TIF1γ |
Skin disease with photoexposed pattern Clinically amyopathic dermatomyositis in younger patients Increased risk of malignancy and muscle weakness in older patients |
Anti-NXP2 |
Subcutaneous calcinosis and edema Severe muscle disease Increased risk of malignancy |
Anti-SAE |
High frequency of dysphagia Skin involvement prior to muscle disease |
