Practice Essentials
Heart failure develops when the heart, via an abnormality of cardiac function (detectable or not), fails to pump blood at a rate commensurate with the requirements of the metabolizing tissues or is able to do so only with an elevated diastolic filling pressure. See the image below.
 Heart Failure. This chest radiograph shows an enlarged cardiac silhouette and edema at the lung bases, signs of acute heart failure.
Heart Failure. This chest radiograph shows an enlarged cardiac silhouette and edema at the lung bases, signs of acute heart failure.
Signs and symptoms
Signs and symptoms of heart failure include the following:
-
Exertional dyspnea and/or dyspnea at rest
-
Orthopnea
-
Acute pulmonary edema
-
Chest pain/pressure and palpitations
-
Tachycardia
-
Fatigue and weakness
-
Nocturia and oliguria
-
Anorexia, weight loss, nausea
-
Exophthalmos and/or visible pulsation of eyes
-
Distention of neck veins
-
Weak, rapid, and thready pulse
-
Rales, wheezing
-
S3 gallop and/or pulsus alternans
-
Increased intensity of P2 heart sound
-
Hepatojugular reflux
-
Ascites, hepatomegaly, and/or anasarca
-
Central or peripheral cyanosis, pallor
See Presentation for more detail.
Diagnosis
Heart failure criteria, classification, and staging
The Framingham criteria for the diagnosis of heart failure consists of the concurrent presence of either two major criteria or one major and two minor criteria. [1]
Major criteria comprise the following:
-
Paroxysmal nocturnal dyspnea
-
Weight loss of 4.5 kg in 5 days in response to treatment
-
Neck vein distention
-
Rales
-
Acute pulmonary edema
-
Hepatojugular reflux
-
S3 gallop
-
Central venous pressure greater than 16 cm water
-
Circulation time of 25 seconds or longer
-
Radiographic cardiomegaly
-
Pulmonary edema, visceral congestion, or cardiomegaly at autopsy
Minor criteria (accepted only if they cannot be attributed to another medical condition) are as follows:
-
Nocturnal cough
-
Dyspnea on ordinary exertion
-
A decrease in vital capacity by one third the maximal value recorded
-
Pleural effusion
-
Tachycardia (rate of 120 bpm)
-
Hepatomegaly
-
Bilateral ankle edema
The New York Heart Association (NYHA) classification system categorizes heart failure on a scale of I to IV, [2] as follows:
-
Class I: No limitation of physical activity
-
Class II: Slight limitation of physical activity
-
Class III: Marked limitation of physical activity
-
Class IV: Symptoms occur even at rest; discomfort with any physical activity
The American College of Cardiology/American Heart Association (ACC/AHA) staging system is defined by the following four stages [3] :
-
Stage A: High risk of heart failure but no structural heart disease or symptoms of heart failure
-
Stage B: Structural heart disease but no symptoms of heart failure
-
Stage C: Structural heart disease and symptoms of heart failure
-
Stage D: Refractory heart failure requiring specialized interventions
Additional ACC/AHA/ and Heart Failure Society of America (HFSA) disease-staging terminology characterizes the syndrome as a continuum [4, 5, 6, 7] :
-
"At risk for HF" for stage A: Applied to asymptomatic patients with risk factors such as diabetes or hypertension but no known cardiac changes
-
"Pre-HF" for stage B: Adds cardiac structural changes or elevated natriuretic peptides, still in the absence of symptoms
-
"Symptomatic HF" for stage C: Structural disease with current or previous symptoms
-
"Advanced HF" for stage D: Characterized by severe debilitating symptoms or repeated hospitalizations even with guideline-directed medical therapy (GDMT)
Testing
The following tests may be useful in the initial evaluation for suspected heart failure [3, 8, 9] :
-
Complete blood cell (CBC) count
-
Iron studies
-
Urinalysis
-
Electrolyte levels
-
Renal and liver function studies
-
Fasting blood glucose levels
-
Lipid profile
-
Thyroid stimulating hormone (TSH) levels
-
B-type natriuretic peptide levels
-
N-terminal pro-B-type natriuretic peptide levels
-
Electrocardiography
-
Chest radiography
-
Two-dimensional (2-D) echocardiography
-
Nuclear imaging [10]
-
Maximal exercise testing
-
Pulse oximetry or arterial blood gas
-
Noninvasive stress testing.
See Workup for more detail.
Management
Treatment includes the following:
-
Nonpharmacologic therapy: Oxygen and noninvasive positive pressure ventilation, dietary sodium and fluid restriction, physical activity as appropriate, and attention to weight gain
-
Pharmacotherapy: Diuretics, vasodilators, inotropic agents, anticoagulants, beta blockers, ACEIs, ARBs, CCBs, digoxin, nitrates, B-type natriuretic peptids, I(f) inhibitors, ARNIs, soluble guanylate cyclase stimulators, SGLT2Is, and MRAs
Surgical options
Surgical treatment options include the following:
-
Electrophysiologic intervention
-
Revascularization procedures
-
Valve replacement/repair
-
Ventricular restoration
-
Extracorporeal membrane oxygenation
-
Ventricular assist devices
-
Heart transplantation
-
Total artificial heart
See Treatment and Medication for more detail.
Background
Heart failure is the pathophysiologic state in which the heart, via an abnormality of cardiac function (detectable or not), fails to pump blood at a rate commensurate with the requirements of the metabolizing tissues or is able to do so only with an elevated diastolic filling pressure.
Heart failure (see the images below) may be caused by myocardial failure but may also occur in the presence of near-normal cardiac function under conditions of high demand. Heart failure always causes circulatory failure, but the converse is not necessarily the case, because various noncardiac conditions (eg, hypovolemic shock, septic shock) can produce circulatory failure in the presence of normal, modestly impaired, or even supranormal cardiac function. To maintain the pumping function of the heart, compensatory mechanisms increase blood volume, cardiac filling pressure, heart rate, and cardiac muscle mass. However, despite these mechanisms, there is a progressive decline in the ability of the heart to contract and relax, resulting in worsening heart failure.
Heart Failure. This chest radiograph shows an enlarged cardiac silhouette and edema at the lung bases, signs of acute heart failure.
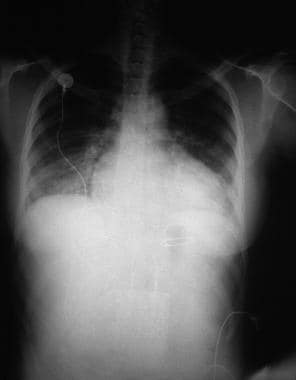 Heart Failure. A 28-year-old woman presented with acute heart failure secondary to chronic hypertension. The enlarged cardiac silhouette on this anteroposterior (AP) radiograph is caused by acute heart failure due to the effects of chronic high blood pressure on the left ventricle. The heart then becomes enlarged, and fluid accumulates in the lungs (ie, pulmonary congestion).
Heart Failure. A 28-year-old woman presented with acute heart failure secondary to chronic hypertension. The enlarged cardiac silhouette on this anteroposterior (AP) radiograph is caused by acute heart failure due to the effects of chronic high blood pressure on the left ventricle. The heart then becomes enlarged, and fluid accumulates in the lungs (ie, pulmonary congestion).
Signs and symptoms of heart failure include tachycardia and manifestations of venous congestion (eg, edema) and low cardiac output (eg, fatigue). Breathlessness is a cardinal symptom of left ventricular (LV) failure that may manifest with progressively increasing severity.
Heart failure can be classified according to a variety of factors (see Heart Failure Criteria, Classification, and Staging). The New York Heart Association (NYHA) classification for heart failure comprises four classes, based on the relationship between symptoms and the amount of effort required to provoke them, as follows [2] :
-
Class I patients have no limitation of physical activity
-
Class II patients have slight limitation of physical activity
-
Class III patients have marked limitation of physical activity
-
Class IV patients have symptoms even at rest and are unable to carry on any physical activity without discomfort
The American College of Cardiology/American Heart Association (ACC/AHA) heart failure guidelines complements the NYHA classification to reflect the progression of disease, divided into four stages, as follows [3] :
-
Stage A patients are at high risk for heart failure but have no structural heart disease or symptoms of heart failure
-
Stage B patients have structural heart disease but have no symptoms of heart failure
-
Stage C patients have structural heart disease and have symptoms of heart failure
-
Stage D patients have refractory heart failure requiring specialized interventions
More recently, the ACC/AHA and Heart Failure Society of America (HFSA) introduced additional disease-staging terminology to characterize the syndrome of heart failure as a continuum, as follows [4, 5, 6, 7] :
-
"At risk for HF" for stage A: Applied to asymptomatic patients with risk factors such as diabetes or hypertension but no known cardiac changes
-
"Pre-HF" for stage B: Adds cardiac structural changes or elevated natriuretic peptides, still in the absence of symptoms
-
"Symptomatic HF" for stage C: Structural disease with current or previous symptoms
-
"Advanced HF" for stage D: Characterized by severe debilitating symptoms or repeated hospitalizations even with guideline-directed medical therapy (GDMT)
Laboratory studies for heart failure should include a complete blood count (CBC), electrolyte levels, and hepatorenal function studies. Imaging studies such as chest radiography and two-dimensional echocardiography are recommended in the initial evaluation of patients with known or suspected heart failure. B-type natriuretic peptide (BNP) and N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels can be useful in differentiating cardiac and noncardiac causes of dyspnea. (See Workup for more information.)
In acute heart failure, patient care consists of stabilizing the patient's clinical condition; establishing the diagnosis, etiology, and precipitating factors; and initiating therapies to provide rapid symptom relief and survival benefit. Surgical options for heart failure include revascularization procedures, electrophysiologic intervention, cardiac resynchronization therapy (CRT), implantable cardioverter-defibrillators (ICDs), valve replacement or repair, ventricular restoration, heart transplantation, and ventricular assist devices (VADs). (See Treatment for more information.)
The goals of pharmacotherapy are to increase survival and to prevent complications. Along with oxygen, medications assisting with symptom relief include diuretics, digoxin, inotropes, and morphine. Drugs that can exacerbate heart failure should be avoided (nonsteroidal anti-inflammatory drugs [NSAIDs], calcium channel blockers [CCBs], and most antiarrhythmic drugs). (See Medication for more information.)
For further information, see the Medscape Drugs & Diseases articles Pediatric Congestive Heart Failure, Congestive Heart Failure Imaging, Heart Transplantation, Pediatric Heart Transplantation, Coronary Artery Bypass Grafting, and Implantable Cardioverter-Defibrillators.
Pathophysiology
The common pathophysiologic state that perpetuates the progression of heart failure is extremely complex, regardless of the precipitating event. Compensatory mechanisms exist on every level of organization, from the subcellular all the way through to organ-to-organ interactions. Only when this network of adaptations becomes overwhelmed does heart failure ensue. [11, 12, 13, 14, 15]
Adaptations
Most important among the adaptations are the following [16] :
-
The Frank-Starling mechanism, in which an increased preload helps to sustain cardiac performance
-
Alterations in myocyte regeneration and death
-
Myocardial hypertrophy with or without cardiac chamber dilatation, in which the mass of contractile tissue is augmented
-
Activation of neurohumoral systems
The release of norepinephrine by adrenergic cardiac nerves augments myocardial contractility and includes activation of the renin-angiotensin-aldosterone system [RAAS], the sympathetic nervous system [SNS], and other neurohumoral adjustments that act to maintain arterial pressure and perfusion of vital organs.
In acute heart failure, the finite adaptive mechanisms that may be adequate to maintain the overall contractile performance of the heart at relatively normal levels become maladaptive when trying to sustain adequate cardiac performance. [17]
The primary myocardial response to chronic increased wall stress is myocyte hypertrophy, death/apoptosis, and regeneration. [18] This process eventually leads to remodeling, usually the eccentric type. Eccentric remodeling further worsens the loading conditions on the remaining myocytes and perpetuates the deleterious cycle. The idea of lowering wall stress to slow the process of remodeling has long been exploited in treating heart failure patients. [19]
The reduction of cardiac output following myocardial injury sets into motion a cascade of hemodynamic and neurohormonal derangements that provoke activation of neuroendocrine systems, most notably the above-mentioned adrenergic systems and RAAS. [20]
The release of epinephrine and norepinephrine, along with the vasoactive substances endothelin-1 (ET-1) and vasopressin, causes vasoconstriction, which increases calcium afterload and, via an increase in cyclic adenosine monophosphate (cAMP), causes an increase in cytosolic calcium entry. The increased calcium entry into the myocytes augments myocardial contractility and impairs myocardial relaxation (lusitropy).
The calcium overload may induce arrhythmias and lead to sudden death. The increase in afterload and myocardial contractility (known as inotropy) and the impairment in myocardial lusitropy lead to an increase in myocardial energy expenditure and a further decrease in cardiac output. The increase in myocardial energy expenditure leads to myocardial cell death/apoptosis, which results in heart failure and further reduction in cardiac output, perpetuating a cycle of further increased neurohumoral stimulation and further adverse hemodynamic and myocardial responses.
In addition, the activation of the RAAS leads to salt and water retention, resulting in increased preload and further increases in myocardial energy expenditure. Increases in renin, mediated by a decreased stretch of the glomerular afferent arteriole, reduce delivery of chloride to the macula densa and increase beta1-adrenergic activity as a response to decreased cardiac output. This results in an increase in angiotensin II (Ang II) levels and, in turn, aldosterone levels, causing stimulation of the release of aldosterone. Ang II, along with ET-1, is crucial in maintaining effective intravascular homeostasis as mediated by vasoconstriction and aldosterone-induced salt and water retention.
The concept of the heart as a self-renewing organ is a relatively recent development. [21] This paradigm for myocyte biology created an entire field of research aimed directly at augmenting myocardial regeneration. The rate of myocyte turnover has been shown to increase during times of pathologic stress. [18] In heart failure, this mechanism for replacement becomes overwhelmed by an even faster increase in the rate of myocyte loss. This imbalance of hypertrophy and death over regeneration is the final common pathway at the cellular level for the progression of remodeling and heart failure.
Angiotensin II
Research indicates that local cardiac Ang II production (which decreases lusitropy, increases inotropy, and increases afterload) leads to increased myocardial energy expenditure. Ang II has also been shown in vitro and in vivo to increase the rate of myocyte apoptosis. [22] In this fashion, Ang II has similar actions to norepinephrine in heart failure.
Ang II also mediates myocardial cellular hypertrophy and may promote progressive loss of myocardial function. The neurohumoral factors above lead to myocyte hypertrophy and interstitial fibrosis, resulting in increased myocardial volume and increased myocardial mass, as well as myocyte loss. As a result, the cardiac architecture changes which, in turn, leads to further increase in myocardial volume and mass.
Myocytes and myocardial remodeling
In the failing heart, increased myocardial volume is characterized by larger myocytes approaching the end of their life cycle. [23] As more myocytes drop out, an increased load is placed on the remaining myocardium, and this unfavorable environment is transmitted to the progenitor cells responsible for replacing lost myocytes.
Progenitor cells become progressively less effective as the underlying pathologic process worsens and myocardial failure accelerates. These features—namely, the increased myocardial volume and mass, along with a net loss of myocytes—are the hallmark of myocardial remodeling. This remodeling process leads to early adaptive mechanisms, such as augmentation of stroke volume (Frank-Starling mechanism) and decreased wall stress (Laplace law) and, later, to maladaptive mechanisms such as increased myocardial oxygen demand, myocardial ischemia, impaired contractility, and arrhythmogenesis.
As heart failure advances, there is a relative decline in the counterregulatory effects of endogenous vasodilators, including nitric oxide (NO), prostaglandins (PGs), bradykinin (BK), atrial natriuretic peptide (ANP), and B-type natriuretic peptide (BNP). This decline occurs simultaneously with the increase in vasoconstrictor substances from the RAAS and the adrenergic system, which fosters further increases in vasoconstriction and thus preload and afterload. This results in cellular proliferation, adverse myocardial remodeling, and antinatriuresis, with total body fluid excess and worsening of heart failure symptoms.
Systolic and diastolic failure
Systolic and diastolic heart failure each result in a decrease in stroke volume. [24, 25] This leads to activation of peripheral and central baroreflexes and chemoreflexes that are capable of eliciting marked increases in sympathetic nerve traffic.
Although there are commonalities in the neurohormonal responses to decreased stroke volume, the neurohormone-mediated events that follow have been most clearly elucidated for individuals with systolic heart failure. The ensuing elevation in plasma norepinephrine directly correlates with the degree of cardiac dysfunction and has significant prognostic implications. Norepinephrine, while directly toxic to cardiac myocytes, is also responsible for a variety of signal-transduction abnormalities, such as downregulation of beta1-adrenergic receptors, uncoupling of beta2-adrenergic receptors, and increased activity of inhibitory G-protein. Changes in beta1-adrenergic receptors result in overexpression and promote myocardial hypertrophy.
Atrial natriuretic peptide and B-type natriuretic peptide
ANP and BNP are endogenously generated peptides activated in response to atrial and ventricular volume/pressure expansion. ANP and BNP are released from the atria and ventricles, respectively, and both promote vasodilation and natriuresis. Their hemodynamic effects are mediated by decreases in ventricular filling pressures, owing to reductions in cardiac preload and afterload. BNP, in particular, produces selective afferent arteriolar vasodilation and inhibits sodium reabsorption in the proximal convoluted tubule. It also inhibits renin and aldosterone release and, therefore, adrenergic activation. ANP and BNP are elevated in chronic heart failure. BNP especially has potentially important diagnostic, therapeutic, and prognostic implications.
For more information, see the Medscape Drugs & Diseases article Natriuretic Peptides in Congestive Heart Failure.
Other vasoactive systems
Other vasoactive systems that play a role in the pathogenesis of heart failure include the ET receptor system, the adenosine receptor system, vasopressin, and tumor necrosis factor-alpha (TNF-alpha). [26] ET, a substance produced by the vascular endothelium, may contribute to the regulation of myocardial function, vascular tone, and peripheral resistance in heart failure. Elevated levels of ET-1 closely correlate with the severity of heart failure. ET-1 is a potent vasoconstrictor and has exaggerated vasoconstrictor effects in the renal vasculature, reducing renal plasma blood flow, glomerular filtration rate (GFR), and sodium excretion.
TNF-alpha has been implicated in response to various infectious and inflammatory conditions. Elevations in TNF-alpha levels have been consistently observed in heart failure and seem to correlate with the degree of myocardial dysfunction. Some studies suggest that local production of TNF-alpha may have toxic effects on the myocardium, thus worsening myocardial systolic and diastolic function.
In individuals with systolic dysfunction, therefore, the neurohormonal responses to decreased stroke volume result in temporary improvement in systolic blood pressure and tissue perfusion. However, in all circumstances, the existing data support the notion that these neurohormonal responses contribute to the progression of myocardial dysfunction in the long term.
Heart failure with preserved ejection fraction
In diastolic heart failure (heart failure with preserved ejection fraction [HFpEF]), the same pathophysiologic processes occur that lead to decreased cardiac output in systolic heart failure, but they do so in response to a different set of hemodynamic and circulatory environmental factors that depress cardiac output. [27]
In HFpEF, altered relaxation and increased stiffness of the ventricle (due to delayed calcium uptake by the myocyte sarcoplasmic reticulum and delayed calcium efflux from the myocyte) occur in response to an increase in ventricular afterload (pressure overload). The impaired relaxation of the ventricle then leads to impaired diastolic filling of the left ventricle (LV).
Morris et al found that right venticular (RV) subendocardial systolic dysfunction and diastolic dysfunction, as detected by echocardiographic strain rate imaging, are common in patients with HFpEF. This dysfunction is potentially associated with the same fibrotic processes that affect the subendocardial layer of the LV and, to a lesser extent, with RV pressure overload. It may play a role in the symptomatology of patients with HFpEF. [28]
LV chamber stiffness
An increase in LV chamber stiffness occurs secondary to any one, or any combination, of the following three mechanisms:
-
Rise in filling pressure
-
Shift to a steeper ventricular pressure-volume curve
-
Decrease in ventricular distensibility
A rise in filling pressure is the movement of the ventricle up along its pressure-volume curve to a steeper portion, as may occur in conditions such as volume overload secondary to acute valvular regurgitation or acute LV failure due to myocarditis.
A shift to a steeper ventricular pressure-volume curve results, most commonly, not only from increased ventricular mass and wall thickness (as observed in aortic stenosis and long-standing hypertension) but also from infiltrative disorders (eg, amyloidosis), endomyocardial fibrosis, and myocardial ischemia.
Parallel upward displacement of the diastolic pressure-volume curve is generally referred to as a decrease in ventricular distensibility. This is usually caused by extrinsic compression of the ventricles.
Concentric LV hypertrophy
Pressure overload that leads to concentric LV hypertrophy (LVH), as occurs in aortic stenosis, hypertension, and hypertrophic cardiomyopathy, shifts the diastolic pressure-volume curve to the left along its volume axis. As a result, ventricular diastolic pressure is abnormally elevated, although chamber stiffness may or may not be altered.
Increases in diastolic pressure lead to an increased myocardial energy expenditure, remodeling of the ventricle, increased myocardial oxygen demand, myocardial ischemia, and eventual progression of the maladaptive mechanisms of the heart that lead to decompensated heart failure.
Arrhythmias
Although life-threatening rhythms are more common in ischemic cardiomyopathy, arrhythmia imparts a significant burden in all forms of heart failure. In fact, some arrhythmias even perpetuate heart failure. The most significant of all rhythms associated with heart failure are the life-threatening ventricular arrhythmias. Structural substrates for ventricular arrhythmias that are common in heart failure, regardless of the underlying cause, include ventricular dilatatation, myocardial hypertrophy, and myocardial fibrosis.
At the cellular level, myocytes may be exposed to increased stretch, wall tension, catecholamines, ischemia, and electrolyte imbalance. The combination of these factors contributes to an increased incidence of arrhythmogenic sudden cardiac death in patients with heart failure.
Etiology
Most patients who present with significant heart failure do so because of an inability to provide adequate cardiac output in that scenario. This is often a combination of the causes listed below in the setting of an abnormal myocardium. The list of causes responsible for presentation of a patient with heart failure exacerbation is very long, and searching for the proximate cause to optimize therapeutic interventions is important.
From a clinical standpoint, classifying the causes of heart failure into the following four broad categories is useful:
-
Underlying causes: Underlying causes of heart failure include structural abnormalities (congenital or acquired) that affect the peripheral and coronary arterial circulation, pericardium, myocardium, or cardiac valves, thus leading to increased hemodynamic burden or myocardial or coronary insufficiency
-
Fundamental causes: Fundamental causes include biochemical and physiologic mechanisms, through which either an increased hemodynamic burden or a reduction in oxygen delivery to the myocardium results in impairment of myocardial contraction
-
Precipitating causes: Overt heart failure may be precipitated by progression of the underlying heart disease (eg, further narrowing of a stenotic aortic valve or mitral valve) or various conditions (fever, anemia, infection) or medications (chemotherapy, nonsteroidal anti-inflammatory drugs [NSAIDs]) that alter the homeostasis of heart failure patients
-
Genetics of cardiomyopathy: Dilated, arrhythmic right ventricular and restrictive cardiomyopathies are known genetic causes of heart failure
Underlying causes
Specific underlying factors cause various forms of heart failure, such as systolic heart failure (most commonly, left vetricular [LV] systolic dysfunction), heart failure with preserved LV ejection fraction (LVEF), acute heart failure, high-output heart failure, and right heart failure.
Underlying causes of systolic heart failure include the following:
-
Coronary artery disease
-
Diabetes mellitus
-
Hypertension
-
Valvular heart disease (stenosis or regurgitant lesions)
-
Arrhythmia (supraventricular or ventricular)
-
Infections and inflammation (myocarditis)
-
Peripartum cardiomyopathy
-
Congenital heart disease
-
Drugs (either recreational, such as alcohol and cocaine, or therapeutic drugs with cardiac side effects, such as doxorubicin)
-
Idiopathic cardiomyopathy
-
Rare conditions (endocrine abnormalities, rheumatologic disease, neuromuscular conditions)
Underlying causes of diastolic heart failure include the following:
-
Coronary artery disease
-
Diabetes mellitus
-
Hypertension
-
Valvular heart disease (aortic stenosis)
-
Hypertrophic cardiomyopathy
-
Restrictive cardiomyopathy (amyloidosis, sarcoidosis)
-
Constrictive pericarditis
Underlying causes of acute heart failure include the following:
-
Acute valvular (mitral or aortic) regurgitation
-
Myocardial infarction (MI)
-
Myocarditis
-
Arrhythmia
-
Drugs (eg, cocaine, calcium channel blockers, or beta-blocker overdose)
-
Sepsis
Underlying causes of high-output heart failure include the following:
-
Anemia
-
Systemic arteriovenous fistulas
-
Hyperthyroidism
-
Beriberi heart disease
-
Paget disease of bone
-
Albright syndrome (fibrous dysplasia)
-
Multiple myeloma
-
Pregnancy
-
Glomerulonephritis
-
Polycythemia vera
-
Carcinoid syndrome
Underlying causes of right heart failure include the following:
-
LV failure
-
Coronary artery disease (ischemia)
-
Pulmonary hypertension
-
Pulmonary valve stenosis
-
Pulmonary embolism
-
Chronic pulmonary disease
-
Neuromuscular disease
Precipitating causes of heart failure
A previously stable, compensated patient may develop heart failure that is clinically apparent for the first time when the intrinsic process has advanced to a critical point, such as with further narrowing of a stenotic aortic valve or mitral valve. Alternatively, decompensation may occur as a result of the failure or exhaustion of the compensatory mechanisms but without any change in the load on the heart in patients with persistent, severe pressure or volume overload. In particular, consider whether the patient has underlying coronary artery disease or valvular heart disease.
The most common cause of decompensation in a previously compensated patient with heart failure is inappropriate reduction in the intensity of treatment, such as dietary sodium restriction, physical activity reduction, or drug regimen reduction. Uncontrolled hypertension is the second most common cause of decompensation, followed closely by cardiac arrhythmias (most commonly, atrial fibrillation). Arrhythmias, particularly ventricular arrhythmias, can be life threatening. Also, patients with one form of underlying heart disease that may be well compensated can develop heart failure when a second form of heart disease ensues. For example, a patient with chronic hypertension and asymptomatic LV hypertrophy (LVH) may be asymptomatic until an MI develops and precipitates heart failure.
Systemic infection or the development of unrelated illness can also lead to heart failure. Systemic infection precipitates heart failure by increasing total metabolism as a consequence of fever, discomfort, and cough, increasing the hemodynamic burden on the heart. Septic shock, in particular, can precipitate heart failure by the release of endotoxin-induced factors that can depress myocardial contractility.
Cardiac infection and inflammation can also endanger the heart. Myocarditis or infective endocarditis may directly impair myocardial function and exacerbate existing heart disease. The anemia, fever, and tachycardia that frequently accompany these processes are also deleterious. In the case of infective endocarditis, the additional valvular damage that ensues may precipitate cardiac decompensation.
Patients with heart failure, particularly when confined to bed, are at high risk of developing pulmonary emboli, which can increase the hemodynamic burden on the right ventricle (RV) by further elevating RV systolic pressure, possibly causing fever, tachypnea, and tachycardia.
Intense, prolonged physical exertion or severe fatigue, such as may result from prolonged travel or emotional crisis, is a relatively common precipitant of cardiac decompensation. The same is true of exposure to severe climate change (ie, the individual comes in contact with a hot, humid environment or a bitterly cold one).
Excessive intake of water and/or salt and the administration of cardiac depressants or drugs that cause salt retention are other factors that can lead to heart failure. At the European Society of Cardiology 2017 Congress, investigators presented a study comprising more than 4630 people that indicated high daily salt intake (>13.7 g) is associated with a substantial increased risk of developing heart failure, independent of other risk factors. [29, 30]
Because of increased myocardial oxygen consumption and demand beyond a critical level, the following high-output states can precipitate the clinical presentation of heart failure:
-
Profound anemia
-
Thyrotoxicosis
-
Myxedema
-
Paget disease of bone
-
Albright syndrome
-
Multiple myeloma
-
Glomerulonephritis
-
Cor pulmonale
-
Polycythemia vera
-
Obesity
-
Carcinoid syndrome
-
Pregnancy
-
Nutritional deficiencies (eg, thiamine deficiency, beriberi)
Longitudinal data from the Framingham Heart Study has suggested that antecedent subclinical LV systolic or diastolic dysfunction is associated with an increased incidence of heart failure, supporting the notion that heart failure is a progressive syndrome. [31, 32] Another analysis of over 36,000 patients undergoing outpatient echocardiography reported that moderate or severe diastolic dysfunction, but not mild diastolic dysfunction, is an independent predictor of mortality. [33]
Genetics of cardiomyopathy
Autosomal dominant inheritance has been demonstrated in dilated cardiomyopathy and in arrhythmic right ventricular cardiomyopathy. Restrictive cardiomyopathies are usually sporadic and associated with the gene for cardiac troponin I. Genetic tests are available at major genetic centers for cardiomyopathies. [34]
In families with a first-degree relative who has been diagnosed with a cardiomyopathy leading to heart failure, the at-risk patient should be screened and followed. [34] The recommended screening consists of an electrocardiogram and an echocardiogram. If the patient has an asymptomatic LV dysfunction, it should be documented and treated. [34]
Epidemiology
United States statistics
According to 2017 American Heart Association (AHA) data, heart failure affects an estimated 6.5 million Americans aged 20 years and older. [35] With improved survival of patients with acute myocardial infarction and with a population that continues to age, heart failure will continue to increase in prominence as a major health problem in the United States. The AHA projects a 46% increase of heart failure prevalence from year 2012 to year 2030, resulting in 8 million or more Americans aged 18 years or older with heart failure. [35]
Despite a more than decade-long decrease (2000-2012) in the the incidence of heart failure–related deaths in the United States, such deaths are on the rise again, particularly among men and non-Hispanic black populations, according to 2000-2014 data (the most recent data available) released by the Centers for Disease Control and Prevention (CDC) in December 2015. [36, 37] The crude rate for heart failure-related deaths decreased from 103.1 deaths per 100,000 population in 2000 to 89.5 in 2009; it then increased to 96.9 in 2014. The age-adjusted rate for heart failure-related deaths decreased from 105.4 deaths per 100,000 standard population in 2000 to 81.4 in 2012; it then increased to 83.4 in 2013 and to 84.0 in 2014. [36] The trend appears to represent a shift from coronary heart disease as the underlying cause of heart failure deaths toward other cardiovascular and noncardiovascular causes, including malignancies, diabetes, chronic lower respiratory diseases, and renal disease.
Analysis of national and regional trends in hospitalization and mortality among Medicare beneficiaries from 1998-2008 showed a relative decline of 29.5% in heart failure hospitalizations [35, 38] ; however, wide variations were noted between states and races, with black men having the slowest rate of decline. A relative decline of 6.6% in mortality was also observed, although the rate was uneven across states. The length of stay decreased from 6.8 days to 6.4 days, despite an overall increase in the comorbid conditions. [38]
Heart failure statistics for the United States are as follows [35] :
-
An estimated one in eight deaths is from heart failure (about 309,000 deaths caused by heart failure each year)
-
Heart failure accounts for 8.5% of cardiovascular-related deaths
-
Approximately 960,000 new cases of heart failure are diagnosed each year
-
The annual incidence of heart failure in patients older than 65 years is 21 per 1,000 population
-
Rehospitalization rates during the 6 months following discharge are as much as 50% [41]
-
In 2012, the estimated total cost of heart failure in the United States was $30.7 billion (68% of which were direct medical costs); by 2030, the total cost is projected to rise to $69.7 billion, a nearly 127% increase.
The incidence and prevalence of heart failure are higher in black persons, Hispanics, Native Americans, and recent immigrants from developing nations, Russia, and the former Soviet republics. The higher prevalence of heart failure in blacks, Hispanics, and Native Americans is directly related to the higher incidence and prevalence of hypertension and diabetes. This problem is particularly exacerbated by a lack of access to health care and by substandard preventive health care available to the most indigent of individuals in these and other groups; in addition, many persons in these groups do not have adequate health insurance.
The higher incidence and prevalence of heart failure in recent immigrants from developing nations are largely due to a lack of prior preventive health care, a lack of treatment, or substandard treatment for common conditions, such as hypertension, diabetes, rheumatic fever, and ischemic heart disease.
Men and women have a similar incidence and prevalence of heart failure. However, many differences remain between men and women with heart failure, such as the following:
-
Whereas the incidence of heart failure in men approximately doubles with each 10-year age increase between 65 and 85 years, it triples for women between ages 65 to 74 years and 75 to 85 years [35]
-
Women tend to develop heart failure later in life than men do
-
Women are more likely than men to have preserved systolic function
-
Women develop depression more commonly than men do
-
Women have signs and symptoms of heart failure similar to those of men, but they are more pronounced in women
-
Women survive longer with heart failure than men do
The prevalence of heart failure increases with age. [40] The prevalence is 1-2% of the population younger than 55 years and increases to a rate of 10% for persons older than 75 years. Nonetheless, heart failure can occur at any age, depending on the cause.
International statistics
Heart failure is a worldwide problem. The most common cause of heart failure in industrialized countries is ischemic cardiomyopathy, with other causes, including Chagas disease and valvular cardiomyopathy, assuming a more important role in developing countries. However, in developing nations that have become more urbanized and more affluent, eating a more processed diet and leading a more sedentary lifestyle have resulted in an increased rate of heart failure, along with increased rates of diabetes and hypertension. This change was illustrated in a population study in Soweto, South Africa, where the community transformed into a more urban and westernized city, followed by an increase in diabetes, hypertension, and heart failure. [42]
In terms of treatment, one study showed few important differences in uptake of key therapies in European countries with widely differing cultures and varying economic status for patients with heart failure. In contrast, studies of sub-Saharan Africa, where healthcare resources are more limited, have shown poor outcomes in specific populations. [43, 44] For example, in some countries, hypertensive heart failure carries a 25% 1-year mortality, and human immunodeficiency virus (HIV)–associated cardiomyopathy generally progresses to death within 100 days of diagnosis in patients who are not treated with antiretroviral drugs.
Although data regarding developing nations are not as robust as studies of Western society, the following trends in developing nations are apparent:
-
Causes tend to be largely nonischemic
-
Patients tend to present at a younger age
-
Outcomes are largely worse where healthcare resources are limited
-
Isolated right heart failure tends to be more prominent, with a variety of causes having been postulated, ranging from tuberculous pericardial disease to lung disease and pollution
Prognosis
In general, the mortality following hospitalization for patients with heart failure is 10.4% at 30 days, 22% at 1 year, and 42.3% at 5 years, despite marked improvement in medical and device therapy. [35, 45, 46, 47, 48, 49]
Mortality is greater than 50% for patients with New York Heart Association (NYHA) class IV, American College of Cardiology/American Heart Association (ACC/AHA) stage D heart failure. Heart failure associated with acute myocardial infarction (MI) has an inpatient mortality of 20-40%; mortality approaches 80% in patients who are also hypotensive (eg, cardiogenic shock). (See Heart Failure Criteria, Classification, and Staging).
Heart failure related to systolic dysfunction has an associated mortality of 50% after 5 years. [24]
Numerous demographic, clinical and biochemical variables have been reported to provide important prognostic value in patients with heart failure, and several predictive models have been developed. [50]
A study by van Diepen et al suggested that patients with heart failure or atrial fibrillation have a significantly higher risk of noncardiac postoperative mortality than patients with coronary artery disease; this risk should be considered even if a minor procedure is planned. [51]
Bursi et al found that among community patients with heart failure, pulmonary artery systolic pressure (PASP), as assessed by Doppler echocardiography, can strongly predict death and can provide incremental and clinically significant prognostic information independent of known outcome predictors. [52]
In the Framingham Offspring Cohort, higher concentrations of galectin-3, a marker of cardiac fibrosis, were associated with an increased risk for incident heart failure (hazard ratio: 1.28 per 1 standard deviation increase in log galectin-3). Galectin-3 was also associated with an increased risk for all-cause mortality (multivariable-adjusted hazard ratio: 1.15). [53]
A more recent, retrospective study that evaluated data from the 2010 Nebraska Hospital Discharge files for 4319 hospitalizations of 3521 heart failure patients admitted to 79 in-state hospitals reported that risk factors for in-hospital mortality in these patients were increasing age, the presence of comordities, and length of hospital day. [54]
In a 2024 prospective, two-center observational study in 350 adults hospitalized for acute heart failure, Espersen et al found four significant independent predictors of the composite of heart failure hospitalization or all-cause mortality in acute heart failure were an admission NT-proBNP (N-terminal pro B-type natriuretic peptide) level of 2000 pg/mL or above, a baseline systolic blood pressure below 120 mmHg, a baseline left atrial volume index of 60 mL/m2 or greater, and 9 or more B-lines on predischarge 4-zone lung ultrasound score (LUS). [55] The investigators noted similar results in a subset of patients (n = 176) with 8-zone LUS data.
Patient Education
To help prevent recurrence of heart failure in patients in whom heart failure was caused by dietary factors or medication noncompliance, counsel and educate such patients about the importance of proper diet and the necessity of medication compliance.
Dunlay et al examined medication use and adherence among community-dwelling patients with heart failure and found that medication adherence was suboptimal in many patients, often because of cost. [56] A randomized controlled trial of 605 patients with heart failure reported that the incidence of all-cause hospitalization or death was not reduced in patients receiving multisession self-care training compared to those receiving a single-session intervention. [57] The optimum method for patient education remains to be established. It appears that more intensive interventions are not necessarily better. [57]
-
Heart Failure. This chest radiograph shows an enlarged cardiac silhouette and edema at the lung bases, signs of acute heart failure.
-
Heart Failure. Cardiac cirrhosis. Congestive hepatopathy with large renal vein.
-
Heart Failure. Cardiac cirrhosis. Congestive hepatopathy with large inferior vena cava.
-
Heart Failure. This electrocardiogram (ECG) is from a 32-year-old female with recent-onset congestive heart failure and syncope. The ECG demonstrates a tachycardia with a 1:1 atrial:ventricular relationship. It is not clear from this tracing whether the atria are driving the ventricles (sinus tachycardia) or the ventricles are driving the atria (ventricular tachycardia [VT]). At first glance, sinus tachycardia in this ECG might be considered with severe conduction disease manifesting as marked first-degree atrioventricular block with left bundle branch block. On closer examination, the ECG morphology gives clues to the actual diagnosis of VT. These clues include the absence of RS complexes in the precordial leads, a QS pattern in V6, and an R wave in aVR. The patient proved to have an incessant VT associated with dilated cardiomyopathy.
-
Heart Failure. This is a posteroanterior view of a right ventricular endocardial activation map during ventricular tachycardia in a patient with a previous septal myocardial infarction. The earliest activation is recorded in red; late activation displays as blue to magenta. Fragmented low-amplitude diastolic local electrocardiograms were recorded adjacent to the earliest (red) breakout area, and local ablation in this scarred zone (red dots) resulted in termination and noninducibility of this previously incessant arrhythmia.
-
Heart Failure. A 28-year-old woman presented with acute heart failure secondary to chronic hypertension. The enlarged cardiac silhouette on this anteroposterior (AP) radiograph is caused by acute heart failure due to the effects of chronic high blood pressure on the left ventricle. The heart then becomes enlarged, and fluid accumulates in the lungs (ie, pulmonary congestion).
-
Heart Failure. Epsilon wave on an electrocardiogram in a patient with arrhythmogenic right ventricular dysplasia (ARVD). ARVD is a congenital cardiomyopathy that is characterized by infiltration of adipose and fibrous tissue into the RV wall and loss of myocardial cells. Primary injuries usually are at the free wall of the RV and right atria, resulting in ventricular and supraventricular arrhythmias. The most significant of all rhythms associated with heart failure are the life-threatening ventricular arrhythmias.
-
Heart Failure. Electrocardiogram depicting ventricular fibrillation in a patient with a left ventricular assist device (LVAD). Ventricular fibrillation is often due to ischemic heart disease and can lead to myocardial infarction and/or sudden death.
-
Heart Failure. The rhythm on this electrocardiogram (ECG) is sinus with borderline PR prolongation. There is evidence of an acute/evolving anterior ischemia/myocardial infarction (MI) superimposed on the left bundle branch block (LBBB)–like pattern. Note the primary T-wave inversions in leads V2-V4, rather than the expected discordant (upright) T waves in the leads with a negative QRS. Although this finding is not particularly sensitive for ischemia/MI with LBBB, such primary T-wave changes are relatively specific. The prominent voltage with left atrial abnormality and leftward axis in concert with the left ventricular intraventricular conduction delay (IVCD) are consistent with underlying left ventricular hypertrophy. This ECG is an example of "bundle branch block plus." Image courtesy of https://ecg.bidmc.harvard.edu.
-
Heart Failure. This electrocardiogram (ECG) shows evidence of severe left ventricular hypertrophy (LVH) with prominent precordial voltage, left atrial abnormality, lateral ST-T abnormalities, and a somewhat leftward QRS axis (–15º). The patient had malignant hypertension with acute heart failure, accounting also for the sinus tachycardia (blood pressure initially 280/180 mmHg). The ST-T changes seen here are nonspecific and could be due to, for example, LVH alone or coronary artery disease. However, the ECG is not consistent with extensive inferolateral myocardial infarction. Image courtesy of https://ecg.bidmc.harvard.edu.
-
Heart Failure. The rhythm on this electrocardiogram is atrial tachycardia (rate, 154 beats/min) with a 2:1 atrioventricular (AV) block. Note the partially hidden, nonconducted P waves on the ST segments (eg, leads I and aVL). The QRS is very wide with an atypical intraventricular conduction defect (IVCD) pattern. The rSR' type complex in the lateral leads (I, aVL) is not due to a right bundle branch block (RBBB) but to an atypical left ventricular conduction defect. These unexpected rSR' complexes in the lateral leads (El-Sherif sign) correlate with underlying extensive myocardial infarction (MI) and, occasionally, ventricular aneurysm. (El-Sherif. Br Heart J. 1970;32:440-8.) The notching on the upstroke of the S waves in lead V4 with a left bundle branch block-type pattern also suggests underlying MI (Cabrera sign). This patient had severe cardiomyopathy secondary to coronary artery disease, with extensive left ventricular wall motion abnormalities. Image courtesy of https://ecg.bidmc.harvard.edu.
-
Heart Failure. On this electrocardiogram, baseline artifact is present, simulating atrial fibrillation. Such artifact may be caused by a variety of factors, including poor electrode contact, muscle tremor, and electrical interference. A single premature ventricular complex (PVC) is present with a compensatory pause such that the RR interval surrounding the PVC is twice as long as the preceding sinus RR interval. Evidence of a previous anterior myocardial infarction is present with pathologic Q waves in leads V1-V3. Borderline-low precordial voltage is a nonspecific finding. Cardiac catheterization showed a 90% stenosis in the patient's proximal portion the left anterior descending coronary artery, which was treated with angioplasty and stenting. Broad P waves in lead V1 with a prominent negative component is consistent with a left atrial abnormality. Image courtesy of https://ecg.bidmc.harvard.edu.
-
Heart Failure. This electrocardiogram (ECG) is from a patient who underwent urgent cardiac catheterization, which revealed diffuse severe coronary spasm (most marked in the left circumflex system) without any fixed obstructive lesions. Severe left ventricular wall motion abnormalities were present, involving the anterior and inferior segments. A question of so-called takotsubo cardiomyopathy (left ventricular apical ballooning syndrome) is also raised (see Bybee et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Int Med 2004:141:858-65). The latter is most often reported in postmenopausal, middle-aged to elderly women in the context of acute emotional stress and may cause ST elevations acutely with subsequent T-wave inversions. A cocaine-induced cardiomyopathy (possibly related to coronary vasospasm) is a consideration but was excluded here. Myocarditis may also be associated with this type of ECG and the cardiomyopathic findings shown here. No fixed obstructive epicardial coronary lesions were detected by coronary arteriography. The findings in this ECG include low-amplitude QRS complexes in the limb leads (with an indeterminate QRS axis), loss of normal precordial R-wave progression (leads V1-V3), and prominent anterior/lateral T-wave inversions. Image courtesy of https://ecg.bidmc.harvard.edu.
-
Heart Failure. This electrocardiogram shows an extensive acute/evolving anterolateral myocardial infarction pattern, with ST-T changes most apparent in leads V2-V6, I, and aVL. Slow R-wave progression is also present in leads V1-V3. The rhythm is borderline sinus tachycardia with a single premature atrial complex (PAC) (fourth beat). Note also the low limb-lead voltage and probable left atrial abnormality. Left ventriculography showed diffuse hypokinesis as well as akinesis of the anterolateral and apical walls, with an ejection fraction estimated at 33%. Image courtesy of https://ecg.bidmc.harvard.edu.
-
Heart Failure. This electrocardiogram shows a patient is having an evolving anteroseptal myocardial infarction secondary to cocaine. There are Q waves in leads V2-V3 with ST-segment elevation in leads V2-V5 associated with T-wave inversion. Also noted are biphasic T waves in the inferior leads. These multiple abnormalities suggest occlusion of a large left anterior descending artery that wraps around the apex of the heart (or multivessel coronary artery disease). Image courtesy of https://ecg.bidmc.harvard.edu.
-
Heart Failure. A color-enhanced angiogram of the left heart shows a plaque-induced obstruction (top center) in a major artery, which can lead to myocardial infarction (MI). MIs can precipitate heart failure.
-
Heart Failure. Emphysema is included in the differential diagnosis of heart failure. In this radiograph, emphysema bubbles are noted in the left lung; these can severely impede breathing capacity.
-
Heart Failure. Cervicocephalic fibromuscular dysplasia (FMD) can lead to complications such as hypertension and chronic kidney failure, which can lead to heart failure. In this color Doppler and spectral Doppler ultrasonographic examination of the left internal carotid artery (ICA) in a patient with cervicocephalic FMD, stenoses of about 70% is seen in the ICA.
-
Heart Failure. Cervicocephalic fibromuscular dysplasia (FMD) can lead to complications such as hypertension and chronic kidney failure, which, in turn, can lead to heart failure. Nodularity in an artery is known as the "string-of-beads sign," and it can be seen this color Doppler ultrasonographic image from a 51-year-old patient with low-grade stenosing FMD of the internal carotid artery (ICA).
-
Heart Failure. Electrocardiogram from a 46-year-old man with long-standing hypertension. Note the left atrial abnormality and left ventricular hypertrophy with strain.
-
Heart Failure. Electrocardiogram from a 46-year-old man with long-standing hypertension. Left atrial abnormality and left ventricular hypertrophy with strain is revealed.
-
Heart Failure. Apical four-chamber echocardiogram in a 37-year-old man with arrhythmogenic right ventricular dysplasia (ARVD), a congenital cardiomyopathy. Note the prominent trabeculae and abnormal wall motion of the dilated RV. ARVD can result in ventricular and supraventricular arrhythmias. The most significant of all rhythms associated with heart failure are the life-threatening ventricular arrhythmias.
-
Heart Failure. Cardiac magnetic resonance image (CMRI), short-axis view. This image shows right ventricular (RV) dilatation, trabucular derangement, aneurysm formation, and dyskinetic free wall in a patient with arrhythmogenic RV dysplasia.
-
Heart Failure. This transthoracic echocardiogram demonstrates severe mitral regurgitation with a heavily calcified mitral valve and prolapse of the posterior leaflet into the left atrium.
-
Heart Failure. Echocardiogram of a patient with severe pulmonic stenosis. This image shows a parasternal short-axis view of a thickened pulmonary valve. Pulmonic stenosis can lead to pulmonary hypertension, which can result in hepatic congestion and in right-sided heart failure.
-
Heart Failure. Echocardiogram of a patient with severe pulmonic stenosis. This image shows a Doppler scan of the peak velocity (5.2 m/s) and gradients (peak 109 mmHg, mean 65 mmHg) across the valve.
-
Heart Failure. Echocardiogram of a patient with severe pulmonic stenosis. This image shows moderately severe pulmonary insufficiency (orange color flow) is also present.
-
Heart Failure. This video is an echocardiogram of a patient with severe pulmonic stenosis. The first segment shows the parasternal short-axis view of the thickened pulmonary valve. The second segment shows the presence of moderate pulmonary insufficiency (orange color flow). AV = aortic valve, PA = pulmonary artery, PI = pulmonary insufficiency, PV = pulmonary valve.
-
Heart Failure. Transesophageal echocardiogram with continuous wave Doppler interrogation across the mitral valve. An increased mean gradient of 16 mmHg is revealed, consistent with severe mitral stenosis.
Tables
- Table 1. Framingham Diagnostic Criteria for Heart Failure
- Table 2. Evidence-Based BNP and NT-proBNP Cutoff Values for Diagnosing HF
- Table 3. 2013 American College of Cardiology Foundation/American Heart Association (ACCF/AHA) Heart Failure Staging System
- Table 4. 2022 ACC/AHA/Heart Failure Society of America (HFSA) Heart Failure Staging System
- Table 5. 2022 ACC/AHA/HFSA Classification of Heart Failure (HF) by Left Ventricular Ejection Fraction (LVEF)
Major Criteria |
Minor Criteria |
Paroxysmal nocturnal dyspnea |
Nocturnal cough |
Weight loss of 4.5 kg in 5 days in response to treatment |
Dyspnea on ordinary exertion |
Neck vein distention |
A decrease in vital capacity by one third the maximal value recorded |
Rales |
Pleural effusion |
Acute pulmonary edema |
Tachycardia (rate of 120 bpm) |
Hepatojugular reflux |
Hepatomegaly |
S3 gallop |
Bilateral ankle edema |
Central venous pressure >16 cm water |
|
Circulation time of ≥25 seconds |
|
Radiographic cardiomegaly |
|
Pulmonary edema, visceral congestion, or cardiomegaly at autopsy |
|
Source: Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol. |
|
Criterion |
BNP, pg/mL |
NT-proBNP, pg/mL |
|||
HF Unlikely (LR-Negative) |
HF Likely (LR-Positive) |
HF Unlikely (LR-Negative) |
HF Likely (LR-Positive) |
||
Age, y |
>17 |
< 100 (0.13)* |
>500 (8.1)* |
- |
- |
>21 |
- |
- |
< 300 (0.02)† |
- |
|
21-50 |
- |
- |
- |
>450 (14)† |
|
50-75 |
- |
- |
- |
>900 (5.0)† |
|
>75 |
- |
- |
- |
>1800 (3.1)† |
|
Estimated GFR, < 60 mL/min |
< 200 (0.13)‡ |
>500 (9.3)‡ |
- |
- |
|
BNP = B-type natriuretic peptide; GRF = glomerular filtration rate; HF = heart failure; LR = likelihood ratio; NPV = negative predictive value; NT-pro-BNP = N-terminal proBNP; PPV = positive predictive value; – = not specifically defined. * Derived from Breathing Not Properly data (1586 emergency department [ED] patients, prevalence of HF = 47%). [66] † Derived from PRIDE data (1256 ED patients, prevalence of HF = 57%). [67, 76] ‡ Derived from subset of Breathing Not Properly data (452 ED patients, prevalence of HF = 49%). [75] |
|||||
Stage |
Description |
Examples |
Notes |
A |
At high risk for heart failure but without structural heart disease or symptoms of heart failure |
Patients with coronary artery disease, hypertension, or diabetes mellitus without impaired left ventricular (LV) function, LV hypertrophy (LVH), or geometric chamber distortion |
Patients with predisposing risk factors for developing heart failure No corresponding New York Heart Association (NYHA) functional classification |
B |
Structural heart disease but without signs/symptoms of heart failure |
Patients who are asymptomatic but who have LVH and/or impaired LV function |
Corresponds with patients with NYHA class I |
C |
Structural heart disease with current or past symptoms of heart failure |
Patients with known structural heart disease and shortness of breath and fatigue, as well as reduced exercise tolerance |
The majority of patients with heart failure are in this stage Corresponds with NYHA classes I, II, III and IV |
D |
Refractory heart failure requiring specialized interventions |
Patients who have marked symptoms at rest despite maximal medical therapy |
Patients in this stage may be eligible to receive mechanical circulatory support, receive continuous inotropic infusions, undergo procedures to facilitate fluid removal, or undergo heart transplantation or other procedures Corresponds with patients with NYHA class IV |
Proposed Terminology |
Stage |
Definition and Criteria |
At risk for HF |
A |
At risk of HF; asymptomatic, no structural heart disease nor cardiac biomarkers of stretch injury (eg, patients with hypertension, atherosclerotic cardiovascular disease, diabetes, metabolic syndrome and obesity, exposure to cardiotoxic agents, genetic variant for cardiomyopathy, or positive family history of cardiomyopathy) |
Pre-HF |
B |
No signs/symptoms of HF and evidence of one of the following: Structural heart disease
Evidence for raised filling pressures by invasive hemodynamic measurements or by noninvasive imaging that suggests elevated filling pressures (eg, Doppler echocardiography) Patients with risk factors and raised levels of B-type natriuretic peptides or persistently elevated cardiac troponin in the absence of competing diagnoses that result in such biomarker elevations (eg, acute coronary syndrome, chronic kidney disease, pulmonary embolus, or myopericarditis) |
Symptomatic HF |
C |
Structural heart disease with current or previous symptoms of HF |
Advanced HF |
D |
Marked HF symptoms that interfere with daily life and with repeated hospitalizations despite attempts to optimize guideline-directed medical therapy |
HF = heart failure |
||
HF Type by LVEF |
Criteria |
HF with reduced EF (HFrEF) |
LVEF ≤40% |
HF with improved EF (HFimpEF) |
Previous LVEF ≤40% and a followup LVEF >40% |
HF with mildly reduced EF (HFmrEF) |
LVEF of 41%-49% Evidence of spontaneous/provokable increased LV filling pressures (eg, elevated natriuretic peptide, noninvasive and invasive hemodynamic measurement) |
HF with preserved EF (HFpEF) |
LVEF ≥50% Evidence of spontaneous/provokable increased LV filling pressures (eg, elevated natriuretic peptide, noninvasive and invasive hemodynamic measurement) |

What would you like to print?
- ‘A Turning Point’ in Heart Failure
- In Heart Failure, Cellular Therapy Is Active
- No Need to Restrict Fluids in Stable Heart Failure
- Facts and Fallacies of Body Composition Analysis in Heart Failure: Is BMI Ready for Replacement?
-
 Jul 18, 2025 This Week in Cardiology Podcast
Jul 18, 2025 This Week in Cardiology Podcast
- Relationship Between Exercise-Induced Cardiac Troponin Elevations and Occult Coronary Atherosclerosis in Middle-Aged Athletes


- Overview
- Presentation
- DDx
- Workup
- Treatment
- Approach Considerations
- Nonpharmacologic Therapy
- Pharmacologic Therapy
- Acute Heart Failure Treatment
- Treatment of Heart Failure with Preserved LVEF
- Treatment of Right Ventricular Heart Failure
- Electrophysiologic Intervention
- Revascularization Procedures
- Valvular Surgery
- Ventricular Restoration
- Extracorporeal Membrane Oxygenation
- Ventricular Assist Devices
- Heart Transplantation
- Total Artificial Heart
- Show All
- Guidelines
- Guidelines Summary
- Screening and Genetic Testing
- Diagnostic Procedures
- Nonpharmacologic Therapy
- Pharmacologic Therapy
- Electrophysiologic Intervention
- Revascularization Procedures
- Valvular Surgery
- Mechanical Circulatory Support Devices
- Heart Transplantation
- Management of Acute Decompensated Heart Failure (ADHF)
- 2024 ISHLT Guidelines on Heart Failure-Related Cardiogenic Shock
- 2024 ESC Guidelines on Management of Dietary Sodium and Fluid Intake in Heart Failure
- Show All
- Medication
- Medication Summary
- Beta-Blockers, Alpha Activity
- Beta-Blockers, Beta-1 Selective
- ACE Inhibitors
- ARBs
- Inotropic Agents
- Vasodilators
- Nitrates
- B-type Natriuretic Peptides
- I(f) Inhibitors
- Angiotensin Receptor-Neprilysin Inhibitors (ARNi)
- Diuretics, Loop
- Diuretics, Thiazide
- Diuretics, Other
- Diuretics, Potassium-Sparing
- Aldosterone Antagonists, Selective
- SGLT2 Inhibitors
- Dual SGLT1/2 Inhibitors
- Soluble Guanylate Cyclase Stimulators
- Alpha/Beta Adrenergic Agonists
- Calcium Channel Blockers
- Anticoagulants, Cardiovascular
- Opioid Analgesics
- Show All
- Media Gallery
- Tables
- References


