Practice Essentials
Rhabdomyosarcoma (RMS) is a malignant soft tissue tumor that is considered to originate from immature cells and myogenic satellite cells that are destined to form striated skeletal muscle. Interestingly, RMS can even originate in locations such as the urinary bladder, where skeletal muscle is not typically found. [1] According to Rubin, it is derived from primitive mesenchyme that retained its capacity for skeletal muscle differentiation. [2]
It is the most common soft tissue sarcoma diagnosed in children, with only a few cases found in adults. [3] RMS of the head and neck is primarily a disease of the first decade of life, and it is the most common soft tissue sarcoma in childhood. Approximately 90% of all cases of RMS are diagnosed in individuals younger than 25 years, and within this group, 60-70% are younger than 10 years. RMS represents 3.5% of all malignancies in children aged 0-14 years, with approximately 350 new cases diagnosed each year. [4] The annual incidence of RMS in the United States is 4.5 cases per 1 million children younger than 14 years.
The disease has little propensity for any particular geographic location or ethnic group. However, people of Asian descent have a slightly lower prevalence than that for Blacks or Whites individuals. The male-to-female ratio is approximately 1.5:1.
See the image below.
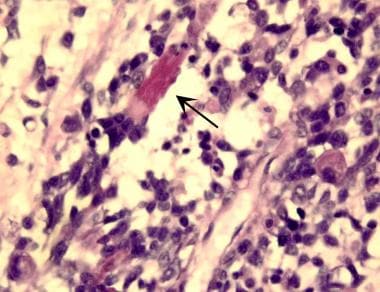 Embryonal rhabdomyosarcoma is evidenced by a variable cell population consisting of small, round tumor cells with hyperchromatic nuclei and of large, polygonal-shaped tumor cells with abundant eosinophilic cytoplasm, which often contains diagnostic cross striations (arrow). (Image provided by Scott Kilpatrick, MD, Department of Pathology, University of North Carolina Hospitals)
Embryonal rhabdomyosarcoma is evidenced by a variable cell population consisting of small, round tumor cells with hyperchromatic nuclei and of large, polygonal-shaped tumor cells with abundant eosinophilic cytoplasm, which often contains diagnostic cross striations (arrow). (Image provided by Scott Kilpatrick, MD, Department of Pathology, University of North Carolina Hospitals)
The head and neck are reportedly the most frequent (35-40%) sites of origin, followed by the genitourinary tract, extremities, trunk, retroperitoneum, and uncommon regions (e.g., intrathoracic, gastrointestinal tract, perianal, and anal regions). However, a retrospective study by Ma et al of pediatric patients with RMS found the genitourinary system to be the most common primary tumor site (43.5%), with the head and neck being the second most common (31.1%) and the extremities the third most common (11.2%); 6.2% of primary tumors were found in the retroperitoneum. [5]
In the head and neck, the most common sites of RMS are parameningeal and orbital locations, which account for 16% and 9% of all cases of the disease, respectively. Over several decades, great progress has been made in the treatment of RMS. As a result, 5-year survival rates increased from 25% in 1970 to 73%, as shown in the Intergroup Rhabdomyosarcoma Study (IRS)-IV reported in 2001. [6]
In a retrospective study of 28 pediatric patients with head and neck RMS published in 2018, Häußler et al found the 5-year overall survival (OS) rate to be 91.3%, with the median period of progression-free survival (PFS) reported to be 46 months. Patients received multimodal treatment, including polychemotherapy (all patients), adjuvant radiation therapy (24 patients), and surgery (12 patients). [7]
In 2006, the National Collaborating Centre for Cancer published a guideline for improving outcomes in patients with sarcoma. [8] Additionally, the National Cancer Institute has provided a patient guide to treatment. [9]
RMS remains a significant pediatric malignancy with variable prognosis depending on histological subtype and tumor location. Early diagnosis and a comprehensive, multimodal treatment approach are critical for improving outcomes in affected patients. Continued research into molecular characterization and targeted therapies holds promise for further advancements in RMS management. [3]
Signs and symptoms of rhabdomyosarcoma
The clinical presentation may vary depending upon the site of the tumor. It is typically asymptomatic, particularly in the initial stages. A mass or area of localized swelling usually characterizes the initial presentation. [3] Fewer than half of these patients present with pain. Pain, along with a rapidly growing mass, is typically seen in pleomorphic RMS. [10]
Other symptoms, which depend on the location of the tumor, include nasal discharge or airway obstruction, otorrhea, hearing loss, fetor (foul smell), and rapid proptosis. Cranial nerve palsies or other neurological deficits indicate extension of the neoplasm into the skull base or central nervous system (CNS).
Workup in rhabdomyosarcoma
The nature and extent of the primary disease must first be determined. To accomplish this, surgical biopsy is performed early in the diagnostic process. In addition, either computed tomography (CT) scanning or magnetic resonance imaging (MRI) is performed to provide accompanying information that is used in planning the approach for surgical resection as well as routes and doses for possible radiation therapy later in the treatment process.
The European guidelines specifically recommend MRI as the method of choice for locoregional evaluation, noting that "CT should not be used for soft tissue tumors if MRI is available." The role of CT in imaging the primary tumor is mainly limited to cases where evaluation of subtle bone destruction is needed. [11]
Next, the clinician must evaluate the patient for locoregional or metastatic disease. This evaluation is accomplished by performing a battery of adjunctive studies, including bone marrow biopsy, chest CT scan, and technetium diphosphonate bone scan. When the primary site is parameningeal, lumbar puncture is also performed for cerebrospinal fluid (CSF) cytology.
Management of rhabdomyosarcoma
Surgery
For superficial, nonorbital lesions in the head and neck, wide excision is recommended. A cuff of normal tissue is always resected with the neoplastic tissue; however, narrow margins are accepted because of anatomic restrictions in the head and neck.
For RMS of the orbit, exenteration is reserved for recurrent or locally persistent disease.
Complete surgical resection with circumferential margins greater than 0.5-1 cm can be done for localized RMS, unless the surgical excision threatens adjacent organs, leads to loss of significant function, results in poor cosmesis, or is not technically feasible. [12]
Primary re-excision is justified if it can be done without important functional or cosmetic sequelae, and if there is a realistic prospect of achieving complete microscopic resection (R0). [12]
If evidence suggests microscopic residual disease after initial surgical resection of head and neck RMS, a second operation with wide margins before chemotherapy may also improve the prognosis.
Chemotherapy
Chemotherapy, in addition to local therapy, has significantly improved the patient outcomes (survival rates in patients with apparently localized disease to approximately 60-90%) in RMS. [13] The most preferred regimen for RMS in North America is a combination of vincristine and dactinomycin (VA protocol) or vincristine, dactinomycin, and cyclophosphamide (VAC protocol), against which other, more or less aggressive combinations have been compared. [14] [13] [15]
Standard treatment for patients with intermediate risk and an intermediate prognosis is the VAC protocol with the addition of radiation therapy. The IRS-V protocol calls for the addition of topotecan to the standard treatment regimen.
Patients with metastatic disease (except the subgroup in the intermediate category) have a survival rate of approximately 30% despite both chemotherapy and irradiation. They are, therefore, considered to be at high risk with a poor prognosis. Like the intermediate group, these patients most commonly receive the VAC protocol. IRS-V recommendations call for the addition of irinotecan in this group.
Radiotherapy
In general, irradiation is good for patients with evidence of gross or microscopic residual disease after resection. Chemotherapy is typically administered for 2-3 months before the start of radiation therapy. Radiation treatment is then administered for approximately 5-6 weeks.
Histology
The current classification of RMS has evolved from the previous five-category system. The most current classification recognizes four major histological categories: embryonal, alveolar, spindle cell/sclerosing, and pleomorphic RMS. The category "sarcoma, not otherwise specified" is used for tumors that could not be classified into a specific subtype and for diffusely anaplastic sarcomas. These tumors were included as pleomorphic sarcomas in older classifications and are associated with a poor prognosis. [16]
Embryonal rhabdomyosarcoma
Embryonal RMS (ERMS) is the most common subtype observed in children, accounting for approximately 60% of all cases in this age group. The tumors can occur at any site, but they are most commonly observed in the genitourinary region or the head and neck region.
Histologically, ERMS shows typical rhabdomyoblasts with moderate to deeply eosinophilic cytoplasm, representing poorly formed myofilaments. The rhabdomyoblasts are arranged in sheets and large nests, with infrequent intermixed fusiform cells and no suggestion of an alveolar architectural pattern. The background can be loose myxoid, or the cells can be compactly arranged in sheets. [16]
On histological examination, they have high cytological variability, which represents several stages of skeletal muscle morphogenesis. As these cells differentiate, they progressively acquire more cytoplasmic eosinophilia and elongate shapes, often described as "tadpole," "strap," and "spider" cells, which are considered evidence of rhabdomyoblastic differentiation. [16] They may range from highly differentiated neoplasms containing rhabdomyoblasts with large amounts of eosinophilic cytoplasm and cross striations similar to those of poorly differentiated tumor cells (see the image below).
Desmin and muscle-specific actin are the typical stains used to identify RMS. Newer staining agents, such as myogenin and MyoD1, are more specific for skeletal muscle than older stains. Desmin and actin stain smooth muscle as well.
Embryonal rhabdomyosarcoma is evidenced by a variable cell population consisting of small, round tumor cells with hyperchromatic nuclei and of large, polygonal-shaped tumor cells with abundant eosinophilic cytoplasm, which often contains diagnostic cross striations (arrow). (Image provided by Scott Kilpatrick, MD, Department of Pathology, University of North Carolina Hospitals)
ERMS has unique molecular characteristics. ERMS cells show a loss of specific genome material from the short arm of chromosome 11. This consistent loss of the material from the 11p15 region may suggest the presence of a tumor suppressor gene, though the actual gene responsible for ERMS is not yet known. Another molecular feature is its lack of gene amplification. In addition, the cellular deoxyribonucleic acid (DNA) content of ERMS is hyperdiploid (1.1-1.8 times normal DNA). [17] Whole chromosome gains with polysomy of chromosome 8 are common, while whole chromosome losses of 10 and 15 are also noted. [16]
Alveolar rhabdomyosarcoma
The alveolar subtype makes up about 31% of all cases of RMS. It is most frequently observed in adolescents and in patients whose primary sites involve the extremities, the trunk, and the perianal and/or perirectal region. Histologically, alveolar RMS (ARMS) are highly cellular malignant neoplasms composed of primitive cells with round nuclei, with fibrovascular septa that separate the tumor cells into discrete nests. These nests contain central clusters of cells with loss of cohesion around the periphery, giving an "alveolar" appearance. Giant cells with rhabdomyoblastic differentiation are common (see the image below). [16] These cells stain intensely with eosinophilic stain. Cross-striated malignant rhabdomyoblasts are observed in 25% of cases, which is less frequent than what is observed with the embryonal form.
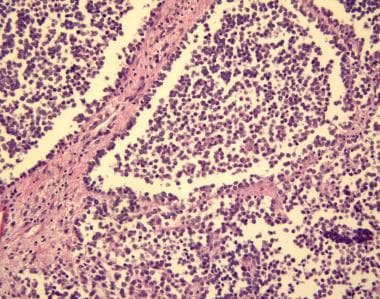 Alveolar rhabdomyosarcoma is evidenced by uniform cell population consisting of cells with a high nuclear-to-cytoplasmic ratio. The cells are arranged in variably sized nests separated by fibrous tissue septa. In places, the cells appear loosely dispersed, mimicking a pulmonary alveolar pattern. (Image provided by Scott Kilpatrick, MD, Department of Pathology, University of North Carolina Hospitals)
Alveolar rhabdomyosarcoma is evidenced by uniform cell population consisting of cells with a high nuclear-to-cytoplasmic ratio. The cells are arranged in variably sized nests separated by fibrous tissue septa. In places, the cells appear loosely dispersed, mimicking a pulmonary alveolar pattern. (Image provided by Scott Kilpatrick, MD, Department of Pathology, University of North Carolina Hospitals)
Immunohistochemically, ARMS stain strongly for desmin. Myogenin and MyoD1 typically show a diffuse, strong nuclear staining pattern. This diffuse positivity for myogenin is unlike the focal patchy staining seen in ERMS and can provide a clue to the subtype. [16]
Like ERMS, ARMS has distinct molecular characteristics. A unique translocation occurs between the FOXO1 gene on chromosome 13 and either the PAX3 gene on chromosome 2 (70%) or the PAX7 gene on chromosome 1 (30%). Individuals with the PAX7 translocation are younger and may have longer event-free survival (EFS) than those with the PAX3 translocation. Unlike ERMS, ARMS commonly demonstrates gene amplification, and its DNA content is typically tetraploidy. [18] [19] [20]
Botryoid rhabdomyosarcoma
Botryoid type, a subset of ERMS, accounts for 6% of all cases of RMS. This subtype characteristically arises under the mucosal surfaces of body orifices; therefore, it is most commonly observed in areas such as the vagina, bladder, and nares. It is distinguished by the formation of polypoid and grapelike tumor masses. On histological study, botryoid RMS demonstrates malignant cells in an abundant myxoid stroma.
The botryoid variant of ERMS contains linear aggregates of tumor cells (cambium layer) that tightly abut an epithelial surface, with variable numbers of polypoid nodules, often with an abundant, loose, myxoid stroma. [16]
Spindle cell/sclerosing rhabdomyosarcoma
This subtype has been reclassified to combine the previously separate spindle cell embryonal and sclerosing RMS categories. [16] The spindle cell subtype of ERMS accounts for 3% of all cases. It has a fascicular, spindled, and leiomyomatous growth pattern and can demonstrate notable rhabdomyoblastic differentiation. Some neoplasms show marked collagen deposition and have a nested, storiform growth pattern. This subtype occurs predominantly in the paratesticular region and is rare in the head and neck. This subset harbors MYOD1 L122R mutations, particularly in adult patients, associated with a more aggressive clinical course. [10]
Pleomorphic rhabdomyosarcoma
Pleomorphic RMS, also known as anaplastic RMS, is the least common subtype and occurs predominantly in adults. [16] It most often occurs in patients aged 30-50 years. It is rarely observed in children. Anaplastic RMS is defined by large, lobate, hyperchromatic nuclei and multipolar mitotic figures.
Presentation and Evaluation
RMS is a disease that can be diagnosed and treated early. Researchers in the IRS-IV reported that 23% of the patients received a diagnosis early enough to receive complete surgical resection, and 15% underwent gross resection with only microscopic residual disease.
Because of the various anatomic constraints of the head and neck, most lesions in this region are obvious at presentation; therefore, they are easily detected. A mass or area of localized swelling usually characterizes the initial presentation. Fewer than half of these patients present with pain.
Other symptoms, which depend on the location of the tumor, include nasal discharge or airway obstruction, otorrhea, hearing loss, fetor (foul smell), and rapid proptosis. Cranial nerve palsies or other neurological deficits indicate extension of the neoplasm into the skull base or CNS. The orbit is involved in nearly one third of head and neck RMSs, followed by those in the oral cavity and oropharynx (29%), face and neck (24%), and middle ear and/or mastoid and sinonasal cavity (9%). Parameningeal sites include the paranasal sinuses, nasal cavity, and middle ear; RMSs at these sites are observed in 16% of patients (see the images below).
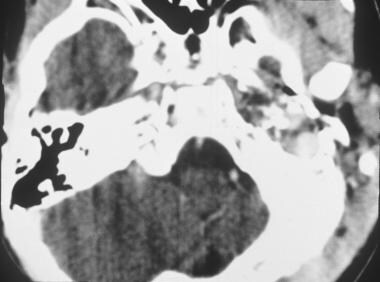 Axial computed tomography scan of rhabdomyosarcoma in the left middle ear. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
Axial computed tomography scan of rhabdomyosarcoma in the left middle ear. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
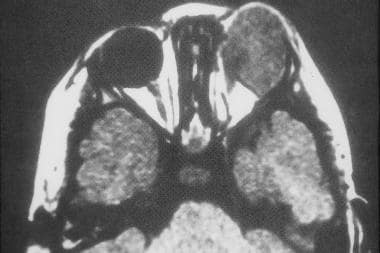 Axial computed tomography scan of left orbital rhabdomyosarcoma. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
Axial computed tomography scan of left orbital rhabdomyosarcoma. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
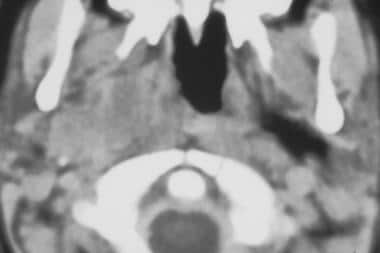 Axial computed tomography scan of right masticator space rhabdomyosarcoma. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
Axial computed tomography scan of right masticator space rhabdomyosarcoma. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
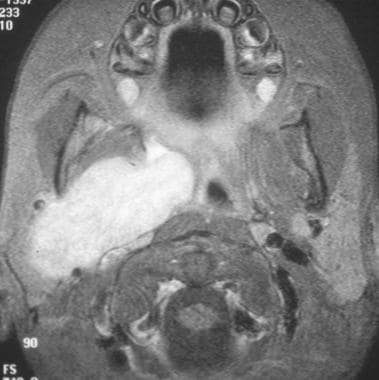 Magnetic resonance imaging of right masticator space rhabdomyosarcoma. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
Magnetic resonance imaging of right masticator space rhabdomyosarcoma. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
Studies have highlighted differences in presentation between pediatric and adult populations. In a 2025 study comparing orbital RMS between these groups, both typically presented with eyelid edema followed by proptosis with similar symptom duration. However, adult patients demonstrated significantly more metastatic disease at presentation, along with increased bone, extraocular muscle, intracranial, and parameningeal involvement. [21]
The initial diagnostic workup must address two key issues. First, the nature and extent of the primary disease must be determined. To accomplish this, surgical biopsy is performed early in the diagnostic process. In addition, either CT scanning or MRI is performed to provide accompanying information that is used in planning the approach for surgical resection as well as routes and doses for possible radiation therapy later in the treatment process.
Prognostic Factors
Multiple clinical and biological factors have been shown to influence the prognosis for a child with RMS. These include the site of tumor origin, tumor size, nodal involvement, histology, and cellular DNA content. Staging classifications based on these factors allow the clinician to determine an overall prognosis for each patient. [22]
The site of origin influences the patient's clinical outcome. For example, patients with head and neck RMS affecting the orbit and nonparameningeal area have a prognosis more favorable than that of patients with tumors in other sites in the body. A study by Affinita et al of pediatric patients with nonparameningeal head and neck RMS reported that the lesions tended to display favorable characteristics, with 72.7% being under 5 cm, 72.7% being classified as T1, and 80.3% being classified as N0. The 10-year overall and PFS rates for the patients — who underwent primary surgery or received chemotherapy along with delayed surgery and/or radiotherapy — were 74.2% and 65.1%, respectively. [23]
Another prognostic factor is tumor burden. Individuals with tumors smaller than 5 cm have an improved prognosis when compared with those with larger tumors. Children with regional nodal involvement do worse than those without nodal disease. Children with metastatic disease have the poorest prognosis. [24] In this group, the most important prognostic factors are histological subtype and the patient's age at diagnosis. For patients who are younger than 10 years and have metastatic disease of embryonal histology, the 5-year survival rate is 60%. Patients older than 10 years with embryonal histology and all patients with alveolar histology have 5-year survival rates of less than 30%. [25]
In a study of 179 cases of RMS (median patient age 14.8 years) in which the disease had metastasized to the bone marrow, Schloemer et al found the rates of EFS at 3 and 5 years to be 9.4% and 8.2%, respectively, with the OS rates being 26.1% and 12.6%, respectively. Among the study's patients, 76% had the alveolar subtype. Poor prognostic/predictive factors included patient age 10 years or older, an alveolar subtype, the presence of FOXO1 fusion, an unfavorable primary site, a greater Oberlin risk factor score, and lack of radiotherapy. [26]
Age is a significant prognostic indicator. Adult patients generally have worse outcomes than pediatric patients, with median PFS of 11 months and median OS of 15 months in one study. [27]
Gender may influence survival in certain subtypes. In sinonasal RSM, male patients exhibited a higher survival rate (hazard ratio = 0.08, 95% CI = 0.01-0.81). [28]
Nutritional status also appears to be prognostically significant in children with RSM; a literature review by Joffe et al found that in the presence of this disease, pediatric patients with an abnormal body mass index (i.e., BMI) trend toward worse OS. [29]
Regional lymph node involvement adversely affects prognosis. Patients presenting with metastatic disease at diagnosis exhibit significantly lower survival rates. Factors such as age at diagnosis, histological subtype, and the number and sites of metastases further stratify risk. Notably, involvement of bone or bone marrow and the presence of three or more metastatic sites are associated with worse outcomes. [30]
The final clinical factor affecting the patient's prognosis is the extent of disease following initial surgical resection. As discussed in Staging Information below, the clinical groups established in the IRS-III and IRS-IV are partially based on the extent of disease after initial surgical resection. Patients without residual disease (group I) have a 90% 5-year survival rate. In patients with microscopic residual disease (group II), survival decreases to 80%, and those with gross disease after surgery (group III) have a 5-year survival rate of 70%.
Biological factors can also influence prognosis. The literature often mentions that the alveolar subtype of RMS is associated with a prognosis worse than that of the other types. When the alveolar subtype is compared with the embryonal type, ARMS is more common in patients with less favorable clinical features (e.g., older age, extremity involvement, distant metastasis). However, when the IRS-I and IRS-II study groups were evaluated, no statistical difference was noted among subtypes when examined independently of other factors.
Cellular DNA content, or ploidy, does appear to have prognostic significance. Patients whose tumor cells have a DNA content 1.5 times higher than normal (hyperdiploid) have a better outcome than those with normal (diploid) or twice-normal (tetraploid) DNA content. Hyperdiploid DNA content is associated with embryonal histology, whereas tetradiploid DNA content is associated with alveolar histology. Also, the incidence of anaplasia in patients with RMS may be higher than previously described and be of prognostic significance in children with intermediate-risk RMS, as its presence appears to negatively influence survival. [31]
In a study of 100 patients under age 21 years (median age 4 years) with large, nonmetastatic primary nongenitourinary ERMS of the abdomen, Dantonello et al concluded that children with these tumors have a fair prognosis if tumor resection or irradiation can be performed after induction chemotherapy. In the study, 36 patients underwent resection following induction chemotherapy, and 60 tumors were irradiated. Median follow-up was 10 years, with patients demonstrating 5-year EFS and OS rates of 52% and 65%, respectively. According to the investigators, significant patient risk factors included age over 10 years, failure to achieve complete remission, and inadequate secondary local therapy (i.e., incomplete secondary resection or absence of radiation therapy). [32]
Fusion status, particularly PAX3::FOXO1 or PAX7::FOXO1, is recognized as a more reliable prognostic indicator than histological classification. Fusion-positive RMS generally has worse outcomes, with a 4-year EFS rate of 53% and OS rate of 69% in localized disease. [33]
Staging Information
Two methods are used to stage RMS. The initial staging system, adopted in the first three intergroup RMS studies, categorizes patients on the basis of the extent of disease and the completeness of their initial surgical resection. Investigators in the IRS-IV attempted to use the tumor, node, and metastasis (TNM) system to standardize the staging system in the United States with those in other parts of the world. Unlike the first system, TNM staging does not take the extent of surgery into account, though it does take size and location into consideration.
The two staging systems are combined, and individuals with RMS are given 1 of 3 risk classifications: low, intermediate, or high. Treatment options are tailored for these risk classifications.
Group staging system
-
Group I - Thirteen percent of all patients with RMS are in group I. This group is defined by localized disease with complete surgical resection and no evidence of regional nodal involvement.
-
Group II - Twenty percent of patients with RMS are in group II.
- Group IIA patients have grossly resected disease with microscopic residual disease and no regional involvement.
- Group IIB patients have had complete resection with no residual disease, but they also have regional disease with involved nodes.
- Group IIC is a hybrid of groups IIA and IIB, containing patients with microscopic residual disease and regional nodal involvement.
-
Group III - Approximately 48% of patients with RMS are in group III. This group is marked by incomplete resection or biopsy only; therefore, it is characterized as gross residual disease.
-
Group IV - Approximately 18% of patients with RMS are in group IV. Individuals in group IV have distant metastasis at the time of diagnosis.
Tumor, node, and metastasis staging system
-
Stage I - Disease is localized and involves the orbit, the head and neck region (excluding parameningeal sites), or the nonbladder and/or nonprostate genitourinary region.
-
Stage II - This stage includes any localized disease of any unfavorable primary site not included in the stage I category. The primary tumor must be less than or equal to 5 cm in diameter.
-
Stage III - The criteria are the same as in stage II, except the primary tumor is larger than 5 cm in diameter and/or it involves regional lymph nodes.
-
Stage IV - Like group IV, stage IV implies metastatic disease at the time of diagnosis.
Risk classification
The most important update is the incorporation of FOXO1 fusion status into risk stratification, which has now replaced histology as a more accurate prognostic factor. Multiple studies have demonstrated that fusion status is a better predictor of outcome than the traditional histological classification of ERMS vs ARMS. This molecular marker has been formally incorporated into the risk stratification systems used by both the Children's Oncology Group (COG) and the European Paediatric Soft Tissue Sarcoma Group. The current COG risk stratification system now integrates FOXO1:PAX fusion status with the traditional staging systems to stratify patients into three main risk groups: [34]
Risk Group |
Stage |
Clinical Group |
Age |
Fusion Status |
Low |
1 |
I, II, III (orbit only) |
Any |
FOXO1 − |
|
2 |
I, II |
Any |
FOXO1 − |
|
|
|
|
|
Intermediate |
1 |
III (non-orbit) |
Any |
FOXO1 − |
|
1, 2, 3 |
I, II, III |
Any |
FOXO1 + |
|
2, 3 |
III |
Any |
FOXO1 − |
|
3 |
I, II |
Any |
FOXO1 − |
|
4 |
IV |
< 10 years |
FOXO1 − |
|
|
|
|
|
High |
4 |
IV |
> 10 years |
FOXO1 − |
|
Any |
FOXO1 + |
Treatment and Management
Treatment of RMS is a multimodality effort. Initial efforts are aimed at surgical resection of the tumor, always followed by chemotherapy and typically ending with a standard course of radiation. The principles of surgical and radiation therapy are based on the site of involvement and the extent of disease, whereas the chemotherapeutic options depend on risk factors.
Surgery
Unlike other regions of the body where RMS can occur, the head and neck region is limited by anatomic constraints; therefore, surgical treatment in this area must be altered accordingly. For superficial, nonorbital lesions, wide excision is recommended. A cuff of normal tissue is always resected with the neoplastic tissue; however, narrow margins are accepted because of the anatomic restrictions.
RMS of the orbit does not require surgical exenteration at the time of initial resection. Initial surgery is aimed only at biopsy for diagnosis. Exenteration is reserved for recurrent or locally persistent disease. If evidence suggests microscopic residual disease after initial surgical resection of head and neck RMS, a second operation with wide margins before chemotherapy may also improve the prognosis.
Surgery plays a limited role in the treatment of metastatic disease. It is indicated only in the context of persistent pulmonary metastases after chemotherapy and irradiation, and it is considered an option only if pulmonary function can be adequately maintained.
Chemotherapy
Risk-factor analysis based on a combination of staging and histology is the primary means for determining the appropriate course of chemotherapy.
Patients at low risk and those with the most favorable prognosis include individuals with ERMS occurring at favorable sites. These include the orbit, nonparameningeal areas of the head and neck, genitourinary nonbladder or nonprostate regions, and biliary tract (stage I). Also included are patients with ERMS at unfavorable sites with completely resected disease (group I) or ERMS at unfavorable sites with microscopic residual disease after resection (group II). For patients with this prognosis, the most common regimen is VA protocol. [14] In certain subpopulations with preexisting renal abnormality that predispose them to nephrotoxicity, cyclophosphamide is often added (i.e., VAC protocol). The OS rate for this group is more than 90%. [35]
Patients with intermediate risk and an intermediate prognosis include those with ERMS at unfavorable sites and gross disease, those who are younger than 10 years and who have metastatic ERMS, and those with nonmetastatic ARMS at any site. Standard treatment for these patients is the VAC protocol with the addition of radiation therapy. The IRS-V protocol calls for the addition of topotecan to the standard treatment regimen. Survival rates at 5 years after diagnosis are 55-70%.
Additional chemotherapeutic agents, including doxorubicin, cisplatin, etoposide, ifosfamide, topotecan, and melphalan, have been tested in various combinations with and without the standard VAC protocol. However, none of these is superior to VAC alone. With high-dose chemotherapy with reconstitution of stem cells, high-risk patients with metastatic disease have a relapse-free survival rate of 19-44%. However, one comparison of this therapy to standard VAC chemotherapy with radiation therapy showed no significant survival advantage to stem cell reconstitution.
Radiation
Radiation therapy helps achieve local control in patients with residual microscopic or gross disease following surgery and chemotherapy (i.e., clinical group II or III disease). The location and extent of disease after surgical management largely determine the doses for radiation treatment. In general, a margin of 2 cm around the tumor and involved nodes is the guideline for the treatment volume.
Chemotherapy is typically administered for 2-3 months before the start of radiation therapy. Radiation treatment is then administered for approximately 5-6 weeks. The only exception to this rule involves patients with parameningeal disease and evidence of meningeal spread. In this circumstance, radiation is started at the time of diagnosis. During radiation treatment, doses of chemotherapy are altered to avoid the use of radiosensitizing agents (e.g., dactinomycin, doxorubicin). Patients with encranial meningeal extension of parameningeal RMS should receive whole-brain irradiation in addition to radiotherapy of the primary tumor.
In general, irradiation is good for patients with evidence of gross or microscopic residual disease after resection. Patients in group I (complete resection) typically do well without irradiation. However, radiation therapy may be of some benefit in patients with alveolar histology, who may otherwise have a less favorable prognosis.
Patients in clinical group II typically receive total radiation doses of 4100 cGy. Individuals in group III receive approximately 5000 cGy. Although this dose is associated with a relapse rate of more than 30%, the long-term toxic effects of increasing the total dose make more aggressive treatments unfeasible. Current investigators are examining hyperfractionization and brachytherapy as alternatives to current treatment methods. In addition, newer studies indicate that improved risk stratification enables decreased therapy intensity for selected patients without compromising survival. [36]
A study by Clement et al indicated that long-term survivors of pediatric head and neck RMS are at a significantly increased risk of developing pituitary dysfunction, owing to treatment with radiation therapy. The study, of 80 survivors of head and neck RMS, reported pituitary dysfunction in 24 of them (30%) after a median 11-year follow-up period, with risk factors for such dysfunction including external beam radiation therapy (EBRT), a parameningeal tumor site, and ERMS subtype. Children treated with the AMORE (ABlative surgery, MOulage technique brachytherapy, and surgical REconstruction) protocol were less likely than those treated with EBRT to develop pituitary dysfunction. [37]
A study by Mattos et al indicated that chemoradiotherapy in children with head and neck RMS can significantly impact dental and craniofacial features. The investigators found 603 dental alterations — most frequently, root shortening — among the study's 27 patients, with 377 such changes (62.5%) appearing in patients aged 5 years or younger. The study also reported the presence of facial asymmetry (74% of patients), reduced facial depth (70.4% of patients), short-sized mandibles (48.4% of patients), and reduced facial height (77.8% of patients). [38]
The SU2C-SARC032 Phase II trial evaluated the efficacy of adding pembrolizumab, a PD-1 inhibitor, to neoadjuvant radiotherapy in patients with high-risk soft tissue sarcomas, including undifferentiated pleomorphic sarcoma and liposarcoma. The study demonstrated a significant improvement in disease-free survival with the addition of pembrolizumab. While RMS was not directly included in this trial, the positive outcomes suggest potential applicability of immunotherapy in RMS treatment protocols, warranting further investigation. [39]
-
Embryonal rhabdomyosarcoma is evidenced by a variable cell population consisting of small, round tumor cells with hyperchromatic nuclei and of large, polygonal-shaped tumor cells with abundant eosinophilic cytoplasm, which often contains diagnostic cross striations (arrow). (Image provided by Scott Kilpatrick, MD, Department of Pathology, University of North Carolina Hospitals)
-
Alveolar rhabdomyosarcoma is evidenced by uniform cell population consisting of cells with a high nuclear-to-cytoplasmic ratio. The cells are arranged in variably sized nests separated by fibrous tissue septa. In places, the cells appear loosely dispersed, mimicking a pulmonary alveolar pattern. (Image provided by Scott Kilpatrick, MD, Department of Pathology, University of North Carolina Hospitals)
-
Axial computed tomography scan of rhabdomyosarcoma in the left middle ear. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
-
Axial computed tomography scan of left orbital rhabdomyosarcoma. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
-
Axial computed tomography scan of right masticator space rhabdomyosarcoma. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
-
Magnetic resonance imaging of right masticator space rhabdomyosarcoma. (Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals)
Tables
Risk Group |
Stage |
Clinical Group |
Age |
Fusion Status |
Low |
1 |
I, II, III (orbit only) |
Any |
FOXO1 − |
|
2 |
I, II |
Any |
FOXO1 − |
|
|
|
|
|
Intermediate |
1 |
III (non-orbit) |
Any |
FOXO1 − |
|
1, 2, 3 |
I, II, III |
Any |
FOXO1 + |
|
2, 3 |
III |
Any |
FOXO1 − |
|
3 |
I, II |
Any |
FOXO1 − |
|
4 |
IV |
< 10 years |
FOXO1 − |
|
|
|
|
|
High |
4 |
IV |
> 10 years |
FOXO1 − |
|
Any |
FOXO1 + |

What would you like to print?
- Radiotherapy Increases Sarcoma Risk in TP53 Breast Cancer
- Rare Cutaneous Sarcoma Incidence Twice as High in Black Individuals vs White Individuals
- FDA Approves First Engineered Cell Therapy for a Solid Tumor
-
 NUT Carcinoma: Diagnostic Challenges, Emerging Treatments, and Clinical Trials
NUT Carcinoma: Diagnostic Challenges, Emerging Treatments, and Clinical Trials
-
Nov 22, 2024 This Week in Cardiology Podcast
-
Genetic Clues to 'Devastating' Sarcoma Diagnosis Uncovered





