Background
Emphysema and chronic bronchitis are airflow-limited states contained within the disease state known as chronic obstructive pulmonary disease (COPD). [1] Just as asthma is no longer grouped with COPD, the current definition of COPD put forth by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) also no longer distinguishes between emphysema and chronic bronchitis. [2]
Emphysema is pathologically defined as an abnormal permanent enlargement of air spaces distal to the terminal bronchioles, accompanied by the destruction of alveolar walls and without obvious fibrosis. [1] This process leads to reduced gas exchange, changes in airway dynamics that impair expiratory airflow, and progressive air trapping. [3] Clinically, the term emphysema is used interchangeably with chronic obstructive pulmonary disease and COPD.
The theory surrounding this definition has been around since the 1950s, with a key concept of irreversibility and/or permanent acinar damage. However, new data posit that increased collagen deposition leads to active fibrosis, which inevitably is associated with breakdown of the lung's elastic framework. An entity known as combined pulmonary fibrosis and emphysema (CPFE) has been shown to exist in a subset of emphysematous patients. [4] This implies an association between fibrosis and the permanence of alveolar damage. The complex mechanism thought to be responsible is the interplay between Notch and Wnt, two signaling pathways playing critical roles in epithelial and mesenchymal precursor cell maintenance and differentiation.
Pathophysiology
Once innate respiratory defenses of the lung's epithelial cell barrier and mucociliary transport system are infiltrated by foreign/invading antigens (noxious cigarette ingredients, for instance), the responding inflammatory immune cells (including polymorphonuclear cells, eosinophils, macrophages, CD4 positive and CD8 positive lymphocytes) transport the antigens to the bronchial associated lymphatic tissue layer (BALT). It is here where the majority of the release of neutrophilic chemotactic factors is thought to occur. Proteolytic enzymes like matrix-metalloproteinases (MMPs) are mainly released by macrophages, which lead to destruction of the lung's epithelial barrier.
Macrophages are found to be 5- to 10-fold higher in the bronchoalveolar lavage fluid of emphysematous patients. [19] Also, along with macrophages, the release of proteases and free radical hydrogen peroxide from neutrophils adds to the epithelial ruination, specifically with emphasis on the basement membrane. This is why neutrophils are thought to be highly important in the pathogenesis of emphysema at the tissue level, a differentiator to the mainly eosinophilic inflammatory response in airways affected by asthma.
After all, the T lymphocytes in the sputum of emphysematous smokers are mainly CD8 positive cells. [20] These cells release chemotactic factors to recruit more cells (pro-inflammatory cytokines that amplify the inflammation) and growth factors that promote structural change. The inflammation is further amplified by oxidative stress and protease production. Oxidants are produced from cigarette smoke and released from inflammatory cells. Proteases are produced by inflammatory, macrophage, and epithelial cells, which fuel bronchiolar edema from an elastin-destroying protease-antiprotease imbalance. This protease-menace is elastase, released by macrophages, and responsible for breakdown of the lung's fragile elastic lamina (of which elastin is a structural protein component). [19] This is believed to be central in the development of emphysema. Peptides from elastin can be detected in increased quantities in patients with emphysema and AAT. [21]
The repair process of airway remodeling further exacerbates emphysema's anatomical derangements with key characters such as vascular endothelial growth factor (VEGF), which is expressed in airway smooth muscle cells and is responsible for neovascularization and expression of increased and possibly abnormal patterns of fibroblastic development. It is these structural changes of mucus hyperplasia, bronchiolar edema, and smooth muscle hypertrophy and fibrosis in smokers' airways that result in the small airways narrowing of less than two millimeters.
AATD lung disease is due to the relative deficiency in the blood and lungs of the alpha-1 antitrypsin (AAT) protein. Although evidence suggests a more complicated cascade of proteolytic and inflammatory factors as the cause of emphysema in AATD, unopposed neutrophil elastase activity within the pulmonary interstitium with resultant connective tissue destruction remains an important contributor to the pathogenesis of emphysema. [22]
Morphology
Pathologically defined as permanent enlargement of airspaces distal to the terminal bronchioles, emphysema creates an environment leading to a dramatic decline in the alveolar surface area available for gas exchange. Loss of individual alveoli with septal wall destruction leads to airflow limitation via two mechanisms. First, loss of alveolar wall results in a decrease in elastic recoil, which subsequently limits airflow. Second, loss of alveolar supporting structures is indirectly responsible for airway narrowing, again limiting airflow. [23]
Though the paradigm for classification continues to evolve, the described morphological pathology of region-specific emphysema remains in three types: [24]
-
Centriacinar (centrilobular)
-
Panacinar (panlobular)
-
Paraseptal
Centriacinar emphysema is the most common type of pulmonary emphysema mainly localized to the proximal respiratory bronchioles with focal destruction and predominantly found in the upper lung zones. The surrounding lung parenchyma is usually normal with untouched distal alveolar ducts and sacs. Also known as centrilobular emphysema, this entity is associated with and closely related to long-standing cigarette smoking and dust inhalation. [25, 26]
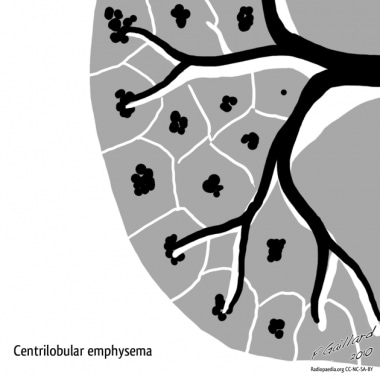 Emphysema. Centrilobular emphysema. Courtesy of Dr Frank Gaillard, Radiopaedia.org (https://radiopaedia.org/cases/emphysema-diagrams).
Emphysema. Centrilobular emphysema. Courtesy of Dr Frank Gaillard, Radiopaedia.org (https://radiopaedia.org/cases/emphysema-diagrams).
Panacinar emphysema destroys the entire alveolus uniformly and is predominant in the lower half of the lungs. Panacinar emphysema generally is observed in patients with homozygous (PiZZ) alpha1-antitrypsin (AAT) deficiency. In people who smoke, focal panacinar emphysema at the lung bases may accompany centriacinar emphysema. [25, 26]
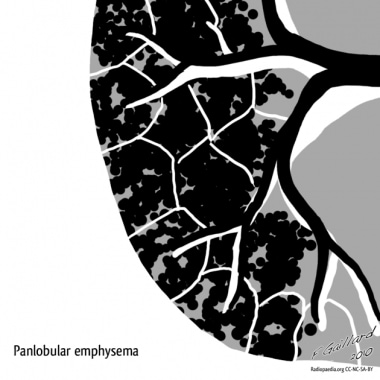 Emphysema. Panlobular emphysema. Courtesy of Dr Frank Gaillard, Radiopaedia.org (https://radiopaedia.org/cases/emphysema-diagrams).
Emphysema. Panlobular emphysema. Courtesy of Dr Frank Gaillard, Radiopaedia.org (https://radiopaedia.org/cases/emphysema-diagrams).
Paraseptal emphysema, also known as distal acinar emphysema, preferentially involves the distal airway structures, alveolar ducts, and alveolar sacs. The process is localized around the septae of the lungs or pleura. Although airflow is frequently preserved, the apical bullae may lead to spontaneous pneumothorax. Giant bullae occasionally cause severe compression of adjacent lung tissue. [25, 26]
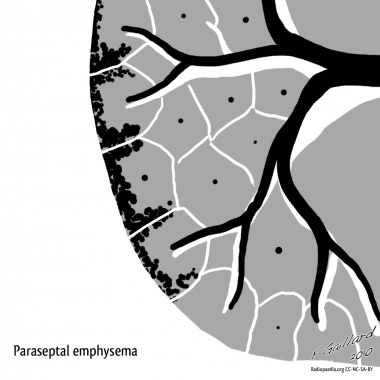 Emphysema. Paraseptal emphysema. Courtesy of Dr Frank Gaillard, Radiopaedia.org (https://radiopaedia.org/cases/emphysema-diagrams).
Emphysema. Paraseptal emphysema. Courtesy of Dr Frank Gaillard, Radiopaedia.org (https://radiopaedia.org/cases/emphysema-diagrams).
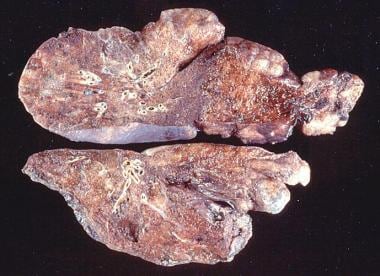
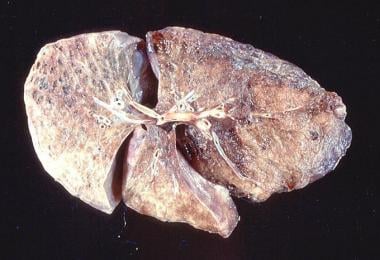 Emphysema. Gross pathology of bullous emphysema shows bullae on the surface of the lungs.
Emphysema. Gross pathology of bullous emphysema shows bullae on the surface of the lungs.
Etiology
Smoking is by far the single most clearly established environmental risk factor for emphysema/chronic bronchitis. Eight out of 10 cases of COPD are caused by smoking. [27] Chronic occupational exposure to inhaled mineral dusts, metal fumes, organic dusts (eg, wood, grains), diesel exhaust fumes, and/or chemical gases or vapors is estimated to be the cause of 19.2% of COPD in smokers and 31.1% of COPD in individuals with no history of smoking. [28, 2]
Alpha-1 antitrypsin deficiency (AATD) is a genetic disorder characterized by the production of an abnormal AAT protein. Although the mechanisms are not completely known, it is believed that in the lungs, low-levels of AAT allow for the destructive effects of neutrophil elastase to go unchecked, which results in damage to the delicate gas exchange region of the lungs (alveoli), eventually leading to emphysema in individuals as young as 30 years of age. Among patients with COPD, up to 3% are estimated to have AATD. It can also cause life threatening liver damage in adults and children and liver cancer in adults. [22]
Emphysema occurs in approximately 2% of persons who use intravenous drugs. This is attributed to pulmonary vascular damage resulting from the insoluble filler (eg, cornstarch, cotton fibers, cellulose, talc) contained in methadone or methylphenidate. The bullous cysts found in association with intravenous use of cocaine or heroin occur predominantly in the upper lobes. In contrast, methadone and methylphenidate injections are associated with basilar and panacinar emphysema.
Human immunodeficiency virus (HIV) infection was found to be an independent risk factor for emphysema, even after controlling for confounding variables such as smoking, intravenous drug use, race, and age. [29] Apical and cortical bullous lung damage occurs in patients who have autoimmune deficiency syndrome and Pneumocystis carinii infection. Reversible pneumatoceles are observed in 10-20% of patients with this infection.
Hypocomplementemic vasculitis urticaria syndrome (HVUS) may be associated with obstructive lung disease. Other sequelae include angioedema, nondeforming arthritis, sinusitis, conjunctivitis, and pericarditis.
Cutis laxa, a disorder of elastin that is characterized most prominently by the appearance of premature aging. The disease is usually congenital, with various forms of inheritance (ie, dominant, recessive). Precocious emphysema has been described in association with cutis laxa as early as the neonatal period or infancy. The pathogenesis of this disorder includes a defect in the synthesis of elastin or tropoelastin.
Marfan syndrome is an autosomal dominant inherited disease of type I collagen characterized by abnormal length of the extremities, subluxation of the lenses, and cardiovascular abnormality. Pulmonary abnormalities, including emphysema, have been described in approximately 10% of patients.
Ehlers-Danlos syndrome refers to a group of inherited connective-tissue disorders with manifestations that include hyperextensibility of the skin and joints, easy bruisability, and pseudotumors.
Salla disease is an autosomal recessive storage disorder described in Scandinavia; the disease is characterized by intralysosomal accumulation of sialic acid in various tissues. The most important clinical manifestations are severe intellectual disability, ataxia, and nystagmus.
Epidemiology
COPD was the third leading cause of death globally and accounted for more than 3.23 million deaths in 2019. [8] In the United States, it is the sixth overall leading cause of death. [7] In 2021, an estimated 14.2 million Americans (6.5%) reported that they were diagnosed with COPD. [9] The prevalence of COPD varies considerably by state, from 3% in Hawaii to 11.8% in West Virginia. Statistically significant increases in COPD prevalence occurred in Colorado, Utah, and West Virginia in the last decade. [9] The states with the highest COPD prevalence are clustered along the Ohio and lower Mississippi Rivers. [30] Because the prevalence is based on the number of adults who have ever been told by any healthcare provider that they have emphysema or chronic bronchitis, the actual number is thought to be much higher. Most patients do not seek medical care until the disease is in its later stages and more than 50% of adults with low pulmonary function were not aware that they had COPD. [10]
The Burden of Obstructive Lung Disease (BOLD) study showed that the global prevalence of COPD (stage II or higher) was 10.1%. [31] This figure varied by geographic location and by sex with a pooled prevalence among men of 11.8% (8.6-22.2%) and among women of 8.5% (5.1-16.7%). The differences can, in part, be explained by site and sex differences in the prevalence of smoking. These rates are similar to rates observed in the Proyecto Latino Americano de Investigacion en Obstruccion Pulmonar (PLATINO study), which studied five countries in Latin America. [32]
The 2014 Surgeon General's report found the risks for COPD were increasing, especially in women. Their risk for COPD is now similar to the risk among men. Women smokers in certain age groups are more than 38 times as likely to develop COPD, compared with women who have never smoked. Moreover, women are dying from COPD more frequently than men, and are more likely to develop severe COPD at younger ages. [27]
Alpha-1 antitrypsin deficiency (AATD) is among the most prevalent potentially fatal genetic disorders in the United States and occurs approximately equally in men and women. The incidence of AADT in the White population is estimated between 1/2500 and 1/3000. Among patients with COPD, up to 3% have AATD. The overwhelming majority of individuals with AATD have not been diagnosed; approximately 10% of the individuals in the United States estimated to have AATD have received a diagnosis. AATD has been identified in virtually all populations but is most common in individuals of Scandinavian, British, Spanish, and Portuguese descent. [22]
Prognosis
Various measures have been shown to correlate with prognosis in COPD, including forced expiratory volume in 1 second (FEV1), diffusion capacity for carbon monoxide (DLCO), blood gas measurements, body mass index (BMI), exercise capacity, and clinical status. A correlation has also been established between radiographic severity of emphysema and mortality. [33]
Patients with features of both asthma and COPD experience more frequent exacerbations, have a poorer quality of life, decline in lung function more rapidly and have a higher mortality rate than patients with COPD alone. [6] Those with COPD and higher serum alpha-1 antitrypsin levels also have a worse systemic inflammation status and higher 10-year mortality. [34]
Comorbidities are common with COPD and have a significant impact on prognosis. Lung cancer is a common cause of death in COPD. Gastroesophageal reflux (GERD) increases the risk of exacerbations. Osteoporosis and depression/anxiety are frequent but under-diagnosed comorbidities that are associated with poor health status and worse prognosis. Other frequent comorbidities include metabolic syndromes, diabetes, cardiovascular disease, hypertension, and bronchiectasis. [2]
A widely used simple prognostication tool is the BODE index, which is based on the BMI, obstruction (FEV1), dyspnea (using Medical Research Council Dyspnea Scale), and exercise capacity (ie, 6-minute walk distance).
BODE index
Body mass index is scored as:
-
Greater than 21 = 0 points
-
Less than 21 = 1 point
FEV1 (postbronchodilator percent predicted) is scored as:
-
Greater than 65% = 0 points
-
50-64% = 1 point
-
36-49% = 2 points
-
Less than 35% = 3 points
The Modified Medical Research Council (mMRC) dyspnea scale is scored as:
-
mMRC 0 = Dyspneic on strenuous exercise (0 points)
-
mMRC 1 = Dyspneic on walking a slight hill (0 points)
-
mMRC 2 = Dyspneic on walking level ground; must stop occasionally due to breathlessness (1 point)
-
mMRC 3 = Dyspneic after walking 100 yards or a few minutes (2 points)
-
mMRC 4 = Cannot leave house; dyspneic doing activities of daily living (3 points)
The 6-minute walking distance is scored as:
-
Greater than 350 meters = 0 points
-
250-349 meters = 1 point
-
150-249 meters = 2 points
-
Less than 149 meters = 3 points
The BODE scoring for approximate 4-year survival is as:
-
0-2 points = 80%
-
3-4 points = 67%
-
5-6 points = 57%
-
7-10 points = 18%
Although age-adjusted death rates for COPD have declined among US men between 1999 (57.0 per 100,000) and 2014 (44.3 per 100,000) in the United States, there has been no significant change among death rates in women (35.3 per 100,000 in 1999 and 35.6 per 100,000 in 2014). Age-adjusted death rates in 2014 varied between states and ranged from 15.3 per 100,000 in Hawaii to 62.8 per 100,000 in Kentucky. The states with the highest COPD death rates are clustered along the Ohio and Mississippi Rivers. [30]
-
Emphysema. Gross pathology of bullous emphysema shows bullae on the surface of the lungs.
-
Emphysema. Gross pathology of emphysema shows bullae on the lung surface.
-
Emphysema. At high magnification, loss of airway walls and dilated airspaces are observed in emphysema.
-
Emphysema. This chest radiograph shows hyperinflation, flattened diaphragms, increased retrosternal space, and hyperlucency of the lung parenchyma in emphysema.
-
Emphysema. A CT scan shows emphysematous bullae in the upper lobes.
-
Emphysema. Diffuse emphysema secondary to cigarette smoking.
-
Emphysema. A pressure-volume curve is drawn for a patient with restrictive lung disease and obstructive disease and is compared to healthy lungs.
-
Emphysema. A flow-volume curve of lungs with emphysema shows a marked decrease in expiratory flows, hyperinflation, and air trapping (patient B) compared to a patient with restrictive lung disease, who has reduced lung volumes and preserved flows (patient A).
-
Emphysema. Forced expiratory volume in 1 second (FEV1) can be used to evaluate the prognosis in patients with emphysema. The benefit of smoking cessation is shown here because the deterioration in lung function parallels that of a nonsmoker, even in late stages of the disease.
-
Emphysema. A CT scan showing severe emphysema and bullous disease.
-
Emphysema. An emphysematous lung shows an increased AP diameter, increased retrosternal airspace, and flattened diaphragms on PA film.
-
Emphysema. An emphysematous lung shows an increased AP diameter, increased retrosternal airspace, and flattened diaphragms on a lateral chest radiograph.
-
Emphysema. The differential diagnosis of a unilateral hyperlucent lung includes pulmonary arterial hypoplasia and Swyer-James syndrome. The expiratory chest radiograph exhibits evidence of air trapping and is helpful in making the diagnosis. Swyer-James syndrome is unilateral bronchiolitis obliterans, which develops during early childhood.
-
Emphysema. A lateral chest radiograph of Swyer-James syndrome may demonstrate some of the features of emphysema.
-
Emphysema. Paraseptal emphysema. Courtesy of Dr Frank Gaillard, Radiopaedia.org (https://radiopaedia.org/cases/emphysema-diagrams).
-
Emphysema. Panlobular emphysema. Courtesy of Dr Frank Gaillard, Radiopaedia.org (https://radiopaedia.org/cases/emphysema-diagrams).
-
Emphysema. Centrilobular emphysema. Courtesy of Dr Frank Gaillard, Radiopaedia.org (https://radiopaedia.org/cases/emphysema-diagrams).
-
Emphysema. Early stethoscope drawing circa 1819. Courtesy of Wikipedia.
-
Emphysema. Laennec's early stethoscope made of brass and wood circa 1820. Courtesy of Wikipedia.
