Practice Essentials
Essential thrombocytosis (primary thrombocythemia) is a nonreactive, chronic myeloproliferative disorder in which sustained megakaryocyte proliferation leads to an increase in the number of circulating platelets [1] (see the images below). Mutations in JAK2, CALR, or MPL are found in approximately 80-90% of patients with essential thrombocytosis. [2] ref52}
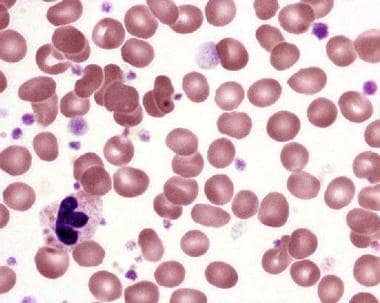 Peripheral blood smear in essential thrombocytosis showing increased platelet numbers. Courtesy Wei Wang, MD, and John Lazarchick, MD; Department of Pathology, Medical University of South Carolina.
Peripheral blood smear in essential thrombocytosis showing increased platelet numbers. Courtesy Wei Wang, MD, and John Lazarchick, MD; Department of Pathology, Medical University of South Carolina.
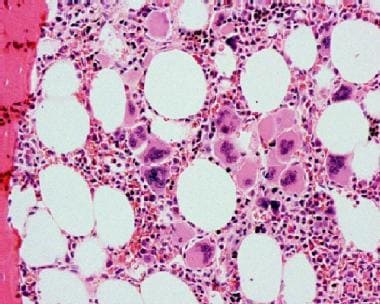 Bone marrow biopsy in essential thrombocytosis showing increased megakaryocytes. Courtesy Wei Wang, MD, and John Lazarchick, MD; Department of Pathology, Medical University of South Carolina.
Bone marrow biopsy in essential thrombocytosis showing increased megakaryocytes. Courtesy Wei Wang, MD, and John Lazarchick, MD; Department of Pathology, Medical University of South Carolina.
Essential thrombocytosis was first described by Epstein and Goedel in 1934, who termed the condition hemorrhagic thrombocythemia. [3, 4] Essential thrombocytosis was traditionally considered a clonal disorder that involved pluripotent stem cells. However, studies have indicated that some patients may have polyclonal hematopoiesis. [5]
Essential thrombocytosis is characterized by the following [6, 7, 8, 9, 10, 11, 12] :
-
A persistently elevated platelet count greater than 450,000/µL
-
Megakaryocytic hyperplasia
-
Splenomegaly
-
A clinical course complicated by thrombotic and/or hemorrhagic episodes
Treatment should be individualized on the basis of risk factors for thrombosis or bleeding. Treatment options range from observation to low-dose aspirin to cytoreductive therapy. In emergencies, plateletpheresis may be useful to achieve a rapid decrease in platelet counts. See Treatment and Medication.
Pathophysiology
Platelet survival is normal in essential thrombocytosis. Instead, the elevated counts result from increased production of platelets by megakaryocytes. The cause of this increase in platelet production remains unclear, but possibilities include the following:
-
Autonomous production
-
Increased sensitivities to cytokines (eg, interleukin-3 [IL-3])
-
Decreased effect of platelet-inhibiting factors (eg, transforming growth factor [TGF] beta)
-
Defects in the accessory cell microenvironment
Bone marrow megakaryocytic precursors (colony-forming unit–megakaryocyte [CFU-Meg]) from patients with essential thrombocytosis form colonies in the absence of exogenous thrombopoietin (TPO). There is no evidence for mutations in the genes for TPO, and patients with essential thrombocytosis have normal or even decreased plasma TPO levels, possibly reflecting increased TPO clearance due to the elevated circulating platelet mass.
The majority of patients with essential thrombocytosis have mutations in one of three genes: Janus kinase 2 (JAK2), calreticulin (CALR), or myeloproliferative leukemia virus oncogene (MPL). These mutations result in the upregulation of the JAK-STAT pathway. Rare cases involve mutations in the thrombopoietin gene (THPO), which are associated with autosomal dominant hereditary thrombocytosis, and somatic mutations in tet methylcytosine dioxygenase 2 (TET2). [13] Most mutations are sporadic; familial cases are thought to be due to an increased propensity for developing mutations as opposed to an inherited germline mutation.
JAK2 mutations possibly turn the thrombopoietin receptor on permanently, leading to overproduction of megakaryocytes. JAK2 mutation is seen in approximately 50-60% of patients.
Somatic mutations in CALR are detected in peripheral blood in the approximately 25% of essential thrombocythemia cases. CALR mutations are mutually exclusive with JAK2 or MPL mutations.
MPL mutations have been associated with only about 3-5% of essential thrombocytosis cases. MPL codes for the thrombopoietin receptor protein, which promotes the growth and proliferation of megakaryocytes. The mutations consist of amino acid substitutions at position 505 in familial cases or 515 in sporadic cases These result in constitutive activation of the thrombopoietin receptor protein. [13]
One study found that patients with JAK2 mutations tend to be older than patients with CALR mutations and to have a higher hemoglobin level and white blood cell count, as well as a lower platelet count and serum erythropoietin level. Risk of thrombosis was twice as high in patients with JAK2 mutations than in those with CALR mutations. Transformation to polycythemia was not observed in patients with CALR mutations, whereas the cumulative risk of polycythemic transformation was 29% at 15 years in those with JAK2 mutations. [14]
The mechanism by which thrombocythemia produces hemorrhage or thrombosis is not well defined. Several defects have been described, including a decrease in aggregation, hyperaggregation, and intracellular concentration of various chemicals. In addition, reports show a decrease in von Willebrand ristocetin cofactor activity and high molecular weight von Willebrand factor multimers. [15, 16] Some reports show patients with an acquired deficiency of antithrombin III, protein C, and protein S. [9]
Etiology
The etiology and predisposing factors for the development of essential thrombocytosis remain unclear. Genetic transmission of this disorder is rare, although reports show several families with multiple members affected. Research suggests that a thrombopoietin production or receptor abnormality can cause familial cases. [17]
Epidemiology
In the United States, clinicians diagnose approximately 6000 cases of essential thrombocytosis each year. Some researchers speculate that the incidence rate may be several times higher. A study from southeastern Minnesota reported an incidence of 2.38 cases per 100,000 population per year. [18]
Essential thrombocytosis is more frequent in older patients. The median age at diagnosis is 60 years, and perhaps up to 20% of patients are younger than 40 years. The disease is rare in children.
In older patients with essential thrombocytosis, the frequency is similar in both sexes. In younger patients, however, essential thrombocytosis occurs more often in women than in men.
In the US, a higher incidence has been noted in Blacks. Lower incidences have been seen in Hispanics and non-Hispanic Whites and Asians/ Pacific Islanders.
Prognosis
The life expectancy of patients with essential thrombocytosis is nearly that of the healthy population. Median survival is approximately 20 years. For patients younger than age 60 years, median survival is 33 years. [11] Unfavorable prognostic factors include advanced age and prior thrombosis. One study reported a 10-year survival rate of 89% and a 15-year survival rate of 80%. [19] These findings were comparable to an age- and sex- standardized European population. Death from essential thrombocytosis–related causes usually occurs from thrombotic complications.
Transformation to acute myelogenous leukemia (AML) occurs in 0.6-5% of patients during the first decade and increases significantly in subsequent decades. [20] Risk factors include advanced age, anemia (hemoglobin less than 12 g/dL in females and less than 13.5 g/dL in males), leukocytosis, platelet count higher than 1,000,000/μL, and sequence variants/mutations involving TP53 and EZH2. [21] Treatment with alkylating agents or radiophosphorus may be associated with higher risk of leukemic transformation.
Morbidity in patients with essential thrombocytosis may involve large-vessel or microvascular thrombosis and bleeding.
-
Peripheral blood smear in essential thrombocytosis showing increased platelet numbers. Courtesy Wei Wang, MD, and John Lazarchick, MD; Department of Pathology, Medical University of South Carolina.
-
Bone marrow biopsy in essential thrombocytosis showing increased megakaryocytes. Courtesy Wei Wang, MD, and John Lazarchick, MD; Department of Pathology, Medical University of South Carolina.
Tables

What would you like to print?
- Cancer Surgery Tied to Increased Venous Thromboembolism Risk
- Maternal Venous Thromboembolism Risk Climbs Steadily: Analysis of Nearly Two Million Pregnancies Raises Concerns
-
 Feb 14, 2025 This Week in Cardiology Podcast
Feb 14, 2025 This Week in Cardiology Podcast
-
Metastatic Endometrial Cancer: Hormonal Therapy Updates and Ways to Target and Treat ER+ Disease
-
Novel Insights in Thrombotic Disorders From ASH 2024