
Overview
Lymphocytic interstitial pneumonia (LIP) is a significant cause of pulmonary pathology in HIV-positive pediatric populations, second only to Pneumocystis infection, and serves as an AIDS-defining condition in approximately 50% of these cases. It is characterized by symptoms such as fever, cough, and dyspnea, accompanied by bibasilar pulmonary infiltrates composed of dense interstitial accumulations of lymphocytes and plasma cells. LIP is rare in adults, affecting less than 1% of individuals, irrespective of HIV status, with a higher prevalence observed in females.
The pathogenesis of LIP may involve autoimmune mechanisms or represent a nonspecific immune response to viral infections such as Epstein-Barr virus, HIV, or other viral agents. The association of LIP with autoimmune and lymphoproliferative disorders—including Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, Hashimoto thyroiditis, myasthenia gravis, pernicious anemia, autoerythrocyte sensitization syndrome, chronic active hepatitis, and common variable immunodeficiency (CVID) [1, 2, 3] —supports an autoimmune etiology. Additionally, LIP is linked to allogeneic bone marrow transplantation, lupus, and lymphoma. Pseudolymphoma represents a localized mass-like variant of LIP, and dysproteinemia has been reported in association with LIP. [4, 5]
Viral etiology is suggested by its frequent occurrence in immunocompromised states, including HIV/AIDS, [6, 7, 8] combined variable immunodeficiency, and agammaglobulinemia. The detection of Epstein-Barr virus DNA, HIV RNA, and human T-cell leukemia virus (HTLV) type 1 in the pulmonary tissue of LIP patients underscores a potential viral influence. Granulomatous lymphocytic interstitial lung disease (GLILD) is a lymphoproliferative complication of CVID, characterized histologically by the presence of both granulomatous and lymphocytic interstitial pneumonitis (LIP), follicular bronchiolitis, and lymphoid hyperplasia. [9]
Diagnostic workup for LIP includes chest radiography, measurement of gas exchange, and histological examination, with laboratory test results generally being nonspecific. Management strategies vary based on symptomatology; asymptomatic and physiologically unaffected patients may not require treatment, whereas symptomatic patients may benefit from supportive care and immunosuppressive therapy, primarily corticosteroids. Cytotoxic therapy has been occasionally employed, and oxygen supplementation may be considered based on blood gas and/or exercise oximetry findings.
For other discussions on pneumonia, see the following:
Pathophysiology and Etiology
HIV-related lymphocytic interstitial pneumonia (LIP) may be part of a continuum of lymphocytic infiltrative disorders, such as pulmonary lymphoid hyperplasia in children and radiographically clear lymphocytic alveolitis in adults. Patients positive for HLA-DR5 and HLA-DR6 alleles are predisposed to developing a diffuse visceral lymphocytosis syndrome with LIP. LIP has been reported to occur as part of immune reconstitution syndrome. [10]
LIP may result from an in situ lymphoproliferative response to chronically presented viral antigens or cytokines and/or recruitment of circulating lymphocytes. Mutations of the B-cell CLL/lymphoma 6 (BCL-6 or zinc finger protein 51) gene have been associated with LIP and mucosa-associated lymphoid tissue (MALT) lymphoma. [11, 12] Viruses (alone or in combination) may be responsible. [13, 14, 15] Potential candidates include Epstein-Barr virus (EBV), human T-lymphotropic virus 1 (HTLV-1), and HIV-1.
Epstein-Barr virus
DNA from EBV is detected in pediatric LIP lung biopsy specimens when accompanied by evidence of primary or reactivated EBV infection at the time of biopsy. Elevated titers of antibodies directed against EBV have been reported in adult patients with LIP.
HTLV-1
HTLV-1 is associated with a spectrum of pulmonary lymphoproliferative syndromes, including LIP. Serologic and molecular studies have correlated HTLV-1 infection with LIP.
The viral transactivating protein p40Tax activates the genes for interleukin-2 (IL-2) and its receptor’s high-affinity alpha chain. Lymphocyte proliferation driven by IL-2 may cause lymphoproliferative pulmonary lesions related to HTLV-1.
HIV-1
The nef gene product induces an LIP-like syndrome in a transgenic mouse model.
Expression of interleukin-18 (IL-18) and IFN-gamma-inducible chemokines IP-10 and Mig is increased in LIP tissues compared with controls. [16] The beta-chemokines RANTES, MIP1-alpha and MIP1-beta, chemotactic for T cells, are increased in pediatric LIP lesions compared with controls. [16]
Infiltrating B cells are polyclonal. Infiltrating T cells in HIV-related LIP are more commonly oligoclonal expansions than in HIV-negative LIP. [17]
BCL-6 mutations in HIV-associated LIP do not show features of immunoglobulin variable heavy chain (IgV[H]) hypermutations, whereas HIV-negative LIP BCL-6 mutations do. The immune dysregulation of HIV-associated LIP appears to be a different type than in HIV-negative LIP.
Enterovirus D68
A case of Enterovirus D68 (EV-D68) infection-caused interstitial pneumonia has been reported. [18] EV-D68 infection is associated with upper and lower respiratory tract symptoms such as fever, cough, and wheezing. Radiologic and pathologic evidence of pneumonia without a bacterial etiology suggests that systemic spread of EV-D68 from a respiratory source may be a component in the pathogenesis of subsequent pneumonia. [19]
Epidemiology
Lymphocytic interstitial pneumonia (LIP) is an uncommon disease. In the United States, however, it is found in 22-75% of pediatric patients with HIV who have pulmonary disease. In contrast, among adult patients with HIV, LIP accounts for only 3% of HIV-related pulmonary pathology. Small series have been reported in Europe, southwestern Japan, Africa, and the Caribbean basin.
Most cases of LIP not associated with HIV occur in the fourth and seventh decades of life, at an average age of 56 years. LIP is common only in children with HIV. In children with HIV infection, lymphocytic interstitial pneumonia has been designated an AIDS-defining illness by the US Centers for Disease Control and Prevention. [20]
LIP is more common in women when not associated with HIV infection. HIV-associated sicca syndrome occurs most often in males. [21]
LIP has been found in every race and HIV risk group. Whether racial or geographic predispositions are crucial remains unclear. Many reports describe HIV and HTLV-1–associated LIP among individuals of African ancestry. [22] LIP appears to cluster in southwestern Japan, where HTLV-1 is endemic.
Prognosis
The clinical course and prognosis of lymphocytic interstitial pneumonia (LIP) in adults are not well-defined due to the rarity of the disease and limited follow-up data. [23] The disease exhibits a variable trajectory, with durations ranging from 1 month to 11 years. LIP may remain stable for months without intervention, improve spontaneously, or recur, occasionally progressing to end-stage fibrosis or bronchiectasis.
Treatment outcomes vary; in non-HIV infected populations, approximately half of the patients respond to treatment with corticosteroids or other immunosuppressive drugs, though relapses are frequent. Despite treatment, some cases may progress to end-stage fibrosis. Historically, older patients have shown higher mortality rates.
The overall 5-year survival rate for LIP ranges from 50% to 66%. [23] Common causes of mortality include infections, the development of malignant lymphoma (5%), and progressive pulmonary fibrosis. In patients with HIV-associated LIP, there is a slower decline in CD4+ T-cell counts and longer survival compared to those with HIV who do not have LIP. [24]
The potential outcomes for LIP include spontaneous resolution, resolution after immunosuppressive therapy, progression to lymphoma, or the development of pulmonary fibrosis leading to respiratory insufficiency. The variability in clinical outcomes underscores the need for further research and detailed patient monitoring.
Complications
Bronchiectasis has been associated with lymphocytic interstitial pneumonia (LIP). Whether this is due to LIP or the frequent bacterial infections these patients experience remains unclear. Bronchitis and pneumonia commonly occur in these patients, with or without bronchiectasis or cystic changes.
Pulmonary fibrosis may be a long-term complication. Generally, it is indolent. Respiratory failure has been reported, especially in the pediatric population.
Malignant transformation to lymphoma or association with lymphoid malignancy has been reported.
Patient Education
Instructions to patients with lymphocytic interstitial pneumonia (LIP) should include relating all potential toxicities of corticosteroids, including aseptic necrosis of the femoral head, infections, weight gain, hyperglycemia, and other adverse effects. Patients should be instructed to seek medical attention for increased dyspnea or change in sputum.
Clinical Presentation
Patient history
In adults, lymphocytic interstitial pneumonia (LIP) typically presents with gradually progressive symptoms, manifesting primarily as dyspnea and chronic cough. [23] These symptoms tend to develop over months or years, with the average onset at around 54 years of age. Although less common, constitutional symptoms such as weight loss, low-grade fever, arthralgias, and night sweats also may occur.
Physical examination may reveal pulmonary crackles. Less frequently observed findings include hepatosplenomegaly, arthritis, and lymphadenopathy, which may indicate an accompanying or alternative diagnosis. Pleuritic chest pain and hemoptysis are rare. Additionally, symptoms of Sicca syndrome, such as xerophthalmia (dry eyes) and xerostomia (dry mouth), may be present. [21]
Physical examination
Manifestations of associated diseases may be present. Physical findings vary in children and adults.
Physical examination findings in children may include the following:
-
Generalized lymphadenopathy
-
Hepatosplenomegaly
-
Parotid enlargement
-
Clubbing
-
Wheezing (occasional)
Physical examination findings in adults may include the following:
-
Generalized lymphadenopathy
-
Rales
-
Hepatosplenomegaly and parotid enlargement: present in approximately one third of adult patients
Differential Diagnosis
The differential diagnosis of LIP includes the following:
Other problems to be considered include the following [25, 26, 27] :
-
Angioimmunoblastic lymphadenopathy
-
Benign lymphocytic angiitis
-
Granulomatosis
-
Nonspecific interstitial pneumonitis
-
Plasma cell interstitial pneumonitis
-
Interstitial lung disease
Laboratory Studies
The diagnosis of lymphocytic interstitial pneumonia (LIP) typically is considered in patients at risk who exhibit compatible clinical symptoms. Diagnostic approaches include imaging studies, laboratory testing, and occasionally, lung biopsy. [23]
A chest X-ray may display bibasilar linear reticular or nodular opacities, which are nonspecific and observed in various pulmonary infections. In more advanced stages of LIP, alveolar opacities and cysts also many be evident.
High-resolution computed tomography (HRCT) of the chest is instrumental in determining the extent of the disease, clarifying the hilar anatomy, and identifying any pleural involvement. HRCT findings can vary widely but often include centrilobular and subpleural nodules, thickened bronchovascular bundles, nodular ground-glass opacities, and cystic structures. Significant hypoxemia may be present in severe cases.
Bronchoalveolar lavage is recommended to exclude infectious etiologies and typically shows an elevated lymphocyte count. Routine laboratory tests, including serum protein electrophoresis (SPEP), are conducted as approximately 80% of patients present with serum protein abnormalities, predominantly polyclonal gammopathy and hypogammaglobulinemia. Serum protein electrophoresis commonly shows polyclonal hypergammaglobulinemia. In pediatric patients with LIP and HIV, lactate dehydrogenase (LDH) levels may be elevated to 300-500 IU/L, approximately half the levels seen in Pneumocystis jiroveci pneumonia; however, this measurement is not helpful in adults. Additional tests such as serologies, immunoglobulin levels, and HIV testing are performed to explore potential secondary causes of LIP, including testing for HIV-1, HTLV-1, EBV, and rheumatoid factor.
For a definitive diagnosis in adults, a lung biopsy is necessary. This biopsy typically reveals expansion of the alveolar septae with infiltrates of lymphocytes and other immune cells such as plasma cells, immunoblasts, and histiocytes. These infiltrates are primarily located along the alveolar septa, though they occasionally may appear along bronchi and vessels. Immunohistochemical staining and flow cytometry are essential for differentiating LIP from primary lymphomas, as LIP is characterized by a polyclonal infiltrate of both T and B cells, in contrast to the monoclonal infiltrates associated with lymphomas. Other histologic features often include germinal centers and multinucleated giant cells within noncaseating granulomas.
Imaging Studies
Chest radiography
Bibasilar interstitial or micronodular infiltrates with coalescence into an alveolar pattern are present in lymphocytic interstitial pneumonia (LIP) (see the image below).
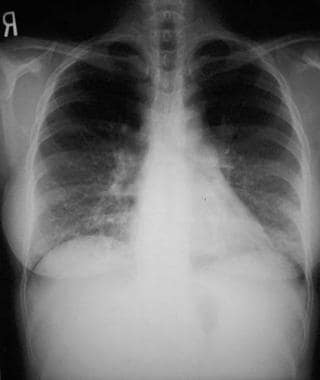 Chest radiograph of lymphocytic interstitial pneumonia in an adult who is HIV positive and has exertional dyspnea, demonstrating characteristic fine bibasilar interstitial markings
Chest radiograph of lymphocytic interstitial pneumonia in an adult who is HIV positive and has exertional dyspnea, demonstrating characteristic fine bibasilar interstitial markings
In adults, honeycombing is present in up to one third of cases. Hilar adenopathy and pleural effusion are uncommon. Similar infiltrates are seen in children, often with mediastinal widening and hilar enlargement denoting pulmonary lymphoid hyperplasia.
Computed tomography
Computed tomography (CT) scanning can distinguish LIP from other diffuse pulmonary diseases and reveal the extent of the disease. CT scanning demonstrates the degree of fibrosis and may demonstrate bronchiectasis. Cyst characteristics, ground-glass attenuation, poorly defined centrilobular nodules, and focal consolidations are associated with LIP. [28, 29, 30]
Findings may be used to follow disease progression. Long-term follow-up may show the development of fibrosis, bronchiectasis, micronodules, bullae, and/or cystic changes. [31, 32]
Other Tests
Arterial blood gas measurement
Arterial blood gas measurement may be helpful in assessing the severity of illness, but the findings are nonspecific.
Partial pressure of oxygen (PO2) measurement is normal. Profound hypoxemia and/or an increased alveolar to arterial (A-a) oxygen gradient is present. Pulse oximetry is used for screening, but it may not detect an A-a gradient. It should be checked at rest and following exercise. See the A-a Gradient calculator.
Pulmonary function testing
Pulmonary function testing usually demonstrates restriction with a reduced or normal diffusion capacity. Obstructive airway disease occasionally has been reported.
Biopsy and Histologic Findings
Generally, bronchoscopy with transbronchial biopsy is diagnostic if multiple biopsies are obtained from several affected subsegments. Exact sensitivity and specificity of transbronchial biopsy is not reported.
Open lung biopsy is the criterion standard. It may be required in the face of nonspecific or equivocal findings, as with extensive fibrosis.
Histology shows alveolar septal and intra-alveolar infiltration by small, mature, noncleaved polyclonal lymphocytes and plasma cells. Lymphoid follicles or micronodules also may be present. No intrapulmonary lymphadenopathy, vasculitis, or necrosis is observed. Extensive areas of interstitial fibrosis may be present. Noncaseating granulomata have been reported.
Treatment and Management
Asymptomatic and physiologically unaffected patients may not require treatment. Symptomatic patients may require supportive care and immunosuppressive treatment, chiefly corticosteroids. [23] Occasionally, cytotoxic therapy has been used. No controlled treatment trials have been reported. [33]
Consultation with a pulmonologist or thoracic surgeon may be necessary to obtain transbronchial biopsy or open lung biopsy, respectively. In cases associated with HIV infection, consultation with a specialist familiar with HIV care is recommended.
In pediatric patients with HIV, empiric treatment for lymphocytic interstitial pneumonia (LIP) often is initiated based on the findings of subacute dyspnea, mild hypoxemia, and clubbing.
Medications should be used in patients who are symptomatic or physiologically compromised.
Corticosteroids
Corticosteroids are used if the patient is symptomatic and/or has physiologic compromise due to LIP. Risks for infection, osteoporosis, hyperglycemia, weight gain, dermatologic changes, and other potential toxicities should be weighed against any potential benefit.
After the first month of therapy and if disease activity allows it, gradually taper the prednisone dosage. Use the lowest possible dose to suppress this chronic interstitial pneumonitis. Monitor the patient for signs of infection and other toxicities of corticosteroid or immunosuppressive therapy.
Immunoglobulin therapy
One report describes dramatic improvement in LIP associated with common variable immunodeficiency treated with intravenous immunoglobulin without steroids. [34]
Immunosuppressive drugs and alkylating agents
The most widely used second-line treatment options are azathioprine, cyclosporine A and cyclophosphamide. [35]
Alkylating agents should be considered only in cases clearly unresponsive to corticosteroids used in high dosage. These agents should only be prescribed by physicians familiar with usage and toxicities. They generally are prescribed for several weeks at a time; disease manifestations and complete blood count should be monitored.
A retrospective chart review of patients with CVID and GLILD found that combination chemotherapy with rituximab and azathioprine resulted in significant improvement in pulmonary function and radiographic lung abnormalities. Comparison of pre- and post-treatment HRCT scans of the chest identified improvements in total score (P=0.018), presence of pulmonary consolidations (P=0.041), ground-glass opacities (P=0.020), nodular opacities (P=0.024), and both the presence and extent of bronchial wall thickening (P=0.014, 0.026 respectively). [9]
Other agents
Antibiotics are used for associated pulmonary infections.
LIP has been reported to improve with the use of zidovudine alone. Highly active antiretroviral therapy (HAART) may result in improvement or resolution of LIP in some instances. [10]
Bronchodilators may be used for associated wheezing.
Oxygen supplementation
Activity may be limited by exercise-induced oxygen desaturation. Perform exercise oximetry to determine if supplementary oxygen is needed. Consider oxygen supplementation based on blood gas and/or exercise oximetry findings.
Long-term monitoring
Periodically perform pulse oximetry at rest and with exercise. Encourage consistent use of a standardized exercise course, such as a long corridor or several flights of steps.
Obtain periodic chest radiographs and/or chest CT scans, which are used for the following purposes:
-
To assess for improvement on therapy
-
To help detect exacerbation of lymphocytic interstitial pneumonia or other pulmonary pathology, notably infections
-
To assess for residual fibrosis
Make every attempt to determine if remaining respiratory compromise is related to pulmonary fibrosis or some other pulmonary pathology.
Obtain clinical reevaluation, radiography, and/or chest CT scan if the patient continues to require high-dose steroids. A change in sputum may be the only sign of infection.
-
Chest radiograph of lymphocytic interstitial pneumonia in an adult who is HIV positive and has exertional dyspnea, demonstrating characteristic fine bibasilar interstitial markings
Tables

What would you like to print?
- Walking Pneumonia Is on the Rise
- New Guidance on Pneumonia Highlight 'Gaps in Knowledge'
- Europe Approves Exblifep for UTIs and Pneumonia
-
 Managing Side Effects and Maximizing Quality of Life in Multiple Myeloma Patients
Managing Side Effects and Maximizing Quality of Life in Multiple Myeloma Patients
-
Jun 27, 2025 This Week in Cardiology Podcast
-
Mar 28, 2025 This Week in Cardiology Podcast






