Definition
Atherosclerosis is an inflammatory condition that arises due to plaque buildup within arterial walls. It is the primary driver in the pathogenesis of coronary, cerebral, and peripheral vascular disease. [1] The term atherosclerosis is derived from the Greek "athero," meaning gruel, or wax, corresponding to the necrotic core area at the base of the atherosclerotic plaque, and "sclerosis" for hardening or induration, referring to the fibrous cap of the plaque's luminal edge. See Coronary Artery Atherosclerosis for information on clinical presentation, differential diagnosis, treatment, and more.
The earliest pathologic descriptions of atherosclerotic lesions focused on morphologies of fatty streaks to fibroatheromas (FAs) and advanced plaques complicated by hemorrhage, calcification, ulceration, and thrombosis. In the mid-1990s the terminology used to define atheromatous plaques was refined by the American Heart Association (AHA) Consensus Group headed by Dr. Stary.
The classification consists of six different numeric categories to include early lesions of initial type I, adaptive intimal thickening; type II, fatty streak; and type III, transitional or intermediate lesions; and advanced plaques characterized as type IV, atheroma; type V, fibroatheroma or atheroma with thick fibrous cap; and type VI, complicated plaques with surface defects and/or hematoma, hemorrhage, and/or thrombosis.
A modified version of the AHA classification was developed to include important pathologic lesions responsible for luminal thrombosis other than plaque rupture, such as plaque erosion and calcified nodule. [2] In this modified classification, numeric AHA lesions types I to IV are replaced by descriptive terminology to include adaptive intimal thickening, intimal xanthoma, pathologic intimal thickening (PIT), and fibroatheroma, as shown in Table 1.
Table 1. Modified AHA Consensus Classification Based on Morphologic Descriptions* (Open Table in a new window)
|
Description |
Thrombosis |
Nonatherosclerotic intimal lesions |
||
Intimal thickening |
Normal accumulation of smooth muscle cells (SMCs) in the intima in the absence of lipid or macrophage foam cells |
Absent |
Intimal xanthoma |
Superficial accumulation of foam cells without a necrotic core or fibrous cap; based on animal and human data, such lesions usually regress |
Absent |
Progressive atherosclerotic lesions |
||
Pathologic intimal thickening |
SMC-rich plaque with proteoglycan matrix and focal accumulation of extracellular lipid |
Absent |
Fibrous cap atheroma |
Early necrosis: focal macrophage infiltration into areas of lipid pools with an overlying fibrous cap Late necrosis: loss of matrix and extensive cellular debris with an overlying fibrous cap |
Absent |
Thin cap fibroatheroma |
A thin, fibrous cap (< 65 µm) infiltrated by macrophages and lymphocytes with rare or absence of SMCs and a relatively large underlying necrotic core; intraplaque hemorrhage/fibrin may be present |
Absent |
Lesions with acute thrombi |
||
Plaque rupture |
Fibroatheroma with fibrous cap disruption; the luminal thrombus communicates with the underlying necrotic core |
Occlusive or nonocclusive |
Plaque erosion |
Plaque composition, as above; no communication of the thrombus with the necrotic core; can occur on a plaque substrate of pathologic intimal thickening or fibroatheroma |
Usually nonocclusive |
Calcified nodule |
Eruptive (shedding) of calcified nodules with an underlying fibrocalcific plaque with minimal or absence of necrosis |
Usually nonocclusive |
Lesions with healed thrombi |
||
Fibrotic (without calcification) Fibrocalcific (+/− necrotic core) |
Collagen-rich plaque with significant luminal stenosis; lesions may contain large areas of calcification with few inflammatory cells and minimal or absence of necrosis; these lesions may represent healed erosions or ruptures |
Absent |
* Modified from Virmani et al [2] Lesion reference to AHA types V and VI was discarded, because it failed to account for the three different morphologies (rupture, erosion, and calcified nodule) that give rise to acute coronary thrombosis. |
||
Etiology
The etiology of atherosclerosis is unknown, but there are multiple factors that contribute to atherosclerotic plaque progression. These include genetic and acquired factors. Processes involved in atherosclerosis include coagulation, inflammation, lipid metabolism, intimal injury, and smooth muscle cell proliferation (see the image below). Genes potentially involved in cardiovascular disease include APOA1, APOA5, APOB, APOC1, APOC2, APOE, CDKN1A, CXCL12, SCARB1, SMARCA4 and TERT. [3]
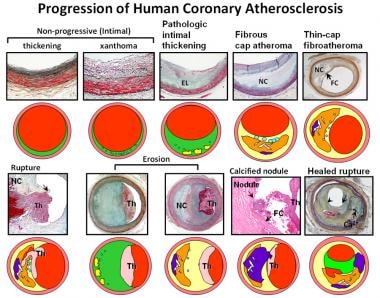 Atherosclerosis pathology. Spectrum of representative coronary lesion morphologies seen in our sudden death population, forming the basis for our modified American Heart Association (AHA) descriptive classification. The two nonprogressive lesions are intimal thickening and intimal xanthomas (foam cell collections known as fatty streaks, AHA type II). Pathologic intimal thickening (PIT) (AHA type III transitional lesions) marks the first of the progressive plaques, as they are the assumed precursors to more advanced fibroatheroma (FA). Thin-cap fibroatheromas (TCFAs) are considered precursors to plaque rupture. Essentially missing from the AHA consensus classification are alternative entities that give rise to coronary thrombosis, namely erosion and the calcified nodule. Erosions can occur on a substrate of PIT or FA, whereas calcified nodules depict eruptive fragments of calcium that protrude into the lumen, causing a thrombotic event. Lastly, healed plaque ruptures are lesions with generally smaller necrotic cores and focal areas of calcification where the surface generally shows areas of healing rich in proteoglycans. Multiple healed plaque ruptures are thought responsible for progressive luminal narrowing. Ca2+ = calcium; EL = extracellular lipid; FC = fibrous cap; NC = necrotic core; Th = luminal thrombus. (Reproduced with permission from Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. May 2000;20(5):1262-75.)
Atherosclerosis pathology. Spectrum of representative coronary lesion morphologies seen in our sudden death population, forming the basis for our modified American Heart Association (AHA) descriptive classification. The two nonprogressive lesions are intimal thickening and intimal xanthomas (foam cell collections known as fatty streaks, AHA type II). Pathologic intimal thickening (PIT) (AHA type III transitional lesions) marks the first of the progressive plaques, as they are the assumed precursors to more advanced fibroatheroma (FA). Thin-cap fibroatheromas (TCFAs) are considered precursors to plaque rupture. Essentially missing from the AHA consensus classification are alternative entities that give rise to coronary thrombosis, namely erosion and the calcified nodule. Erosions can occur on a substrate of PIT or FA, whereas calcified nodules depict eruptive fragments of calcium that protrude into the lumen, causing a thrombotic event. Lastly, healed plaque ruptures are lesions with generally smaller necrotic cores and focal areas of calcification where the surface generally shows areas of healing rich in proteoglycans. Multiple healed plaque ruptures are thought responsible for progressive luminal narrowing. Ca2+ = calcium; EL = extracellular lipid; FC = fibrous cap; NC = necrotic core; Th = luminal thrombus. (Reproduced with permission from Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. May 2000;20(5):1262-75.)
Factors that affect these processes may inhibit or accelerate atherosclerosis. The most common risk factors are family history, hyperlipidemia, diabetes mellitus, cigarette smoking, hypertension, and dietary deficiencies of antioxidants. [4] Early lesion development is marked by lipid retention with activation of endothelial adhesion molecules. Inflammatory macrophages play a significant role throughout all phases of atherosclerotic progression; hyperlipidemia-induced macrophage infiltration of the arterial intima is one of the earliest pathologic changes.
A major event in atherosclerotic plaque progression is thrombosis, which may occur in any arterial bed (coronary, aorta, carotid, etc.) Three different morphologies (rupture, erosion, and calcified nodule) may give rise to acute coronary thrombosis. The development of plaque and its rupture are hallmarks of atherosclerotic vascular disease. [5] Plaque rupture is defined by fibrous cap disruption or fracture, whereby the overlying thrombus is in continuity with the underlying necrotic core. Plaque erosion is identified when serial sectioning through a thrombus fails to show communication with a necrotic core or deep intima; the endothelium is absent, and the thrombus is superimposed on a plaque substrate primarily composed of smooth muscle cells and proteoglycans. Calcified nodules are characterized by eruptive dense calcified bodies protruding into the luminal space and represent the least frequent morphology associated with luminal thrombosis (see the following diagram).
 Atherosclerosis pathology. Simplified scheme for classifying atherosclerotic lesions modified from the current American Heart Association (AHA) recommendations. The boxed areas represent the seven categories of lesions. Dashed lines are used for two categories, because there is controversy over the role that each plays in the initial phase of lesion formation, and both "lesions" can exist without progressing to a fibrous cap atheroma (ie, AHA type IV lesion). The processes responsible for progression are listed between categories. Lines (solid and dotted; dotted lines represent the least-established processes) depict current concepts of how one category may progress to another, with the thickness of the line representing the strength of supportive evidence that the events occur. (Reproduced with permission from Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. May 2000;20(5):1262-75.)
Atherosclerosis pathology. Simplified scheme for classifying atherosclerotic lesions modified from the current American Heart Association (AHA) recommendations. The boxed areas represent the seven categories of lesions. Dashed lines are used for two categories, because there is controversy over the role that each plays in the initial phase of lesion formation, and both "lesions" can exist without progressing to a fibrous cap atheroma (ie, AHA type IV lesion). The processes responsible for progression are listed between categories. Lines (solid and dotted; dotted lines represent the least-established processes) depict current concepts of how one category may progress to another, with the thickness of the line representing the strength of supportive evidence that the events occur. (Reproduced with permission from Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. May 2000;20(5):1262-75.)
A long-term study that began in 1980 to investigate if low vitamin D levels in childhood are related with increased carotid artery intima-media thickness (IMT) in adulthood reported that vitamin D deficiency in childhood may be linked to hardening of the arteries in middle age. [6, 7]
Epidemiology
Despite advances in medical, interventional, and surgical treatment, atherosclerotic disease remains the most important cause of death in developed and underprivileged nations. [8, 9]
In the United States, coronary heart disease accounted for 375,476 deaths in 2021. Each year, a substantial number of Americans experience a myocardial infarction (MI), with 605,000 being first-time events and 200,000 recurrent episodes. [10]
Coronary artery disease remains the leading cause of death in the Western world. A new or recurrent MI afflicts approximately 1.1 million people in the USA per year, of which 40% are fatal.
Sudden cardiac death as a first manifestation of the atherosclerotic process occurs in over 450,000 individuals annually. The vast majority of acute MIs (approximately 75%) occur from plaque rupture; other causes of coronary thrombosis include erosion and calcified nodules. [11]
Although lesions with rupture occur in males of all ages (this is consistent for all plaque morphologies with thrombi), the frequency of sudden coronary death decreases with advancing age. The incidence of rupture varies with each decade, and the highest incidence of plaque rupture is seen in the 40s in males, whereas in females the incidence increases beyond the age of 50 years. Approximately 80% of coronary thrombi in females older than 50 years occur from plaque rupture, and there is a strong association with circulating cholesterol. In acute MI or sudden coronary death, plaque erosion occurs primarily in patients younger than 50 years and represents the majority of acute coronary thrombi in premenopausal females. Furthermore, 20-25% of acute myocardial infarcts occurring in hospitalized patients are due to plaque erosion.
Location
Atherosclerosis occurs in elastic and muscular arteries and may occur iatrogenically in vein grafts interposed in the arterial circulation. The aorta is affected earliest, followed by the carotid arteries, coronary arteries, and iliofemoral arteries. Sawabe et al. have shown that disease progression occurred more rapidly in the aorta, followed by coronary and femoral, and occurred least in the carotid and intracerebral arteries. [12] Initially, lesions are most common at branch points, at sites of low shear, where a predilection to plaque formation has been observed. Coronary lesions, including thrombi occurring at atherosclerotic sites, are most prevalent in the proximal coronary arteries: the proximal left anterior descending coronary artery, followed by the right and left circumflex coronary arteries.
Clinical Features and Imaging
Atherosclerosis causes symptoms by arterial obstruction, embolization of plaque material, and weakening with rupture of the arterial wall. Obstruction with or without embolization causes ischemia of the circulation supplied by the vessel. Ischemic strokes result from atherosclerosis of the carotid arteries and aortic arch, which embolize thrombi and atherosclerotic material, as well as local atherosclerosis of the cerebral vessels.
Obstruction of coronary arteries causes myocardial ischemia. Myocardial ischemia may present as acute coronary syndromes (acute ST elevation infarct, non-ST elevation infarct, and unstable angina), sudden death, or chronic congestive heart failure. Obstruction of iliac vessels results in ischemia of the lower extremities (claudication). Atherosclerotic aneurysms show a predilection for the aorta, especially the abdominal aorta. Aortic aneurysms may rupture and cause death by hemorrhage into the retroperitoneal space or pleural cavities, depending on the location.
The criterion standard for imaging atherosclerotic lesions of the coronary circulation is angiography, whereas computed tomography (CT) angiography is emerging as a promising approach for the noninvasive assessment of coronary artery stenosis and plaque characteristics. [13, 14] Motoyama et al. have reported that the presence of low attenuated plaque and positive remodeling by CT angiography are predictive of future acute coronary events. [15] Advanced imaging modalities, such as cardiac MRI, and positron emission tomography/CT (PET/CT) are being developed that may provide more detailed information besides being less invasive methods of determining sites of stenosis. Imaging of atherosclerotic lesions of the carotid circulation include carotid ultrasonography, a noninvasive technique. [16]
Advancements in MRI techniques allow for detailed visualization of plaque morphology and composition without radiation exposure. MRI can assess features such as intraplaque hemorrhage and lipid-rich necrotic cores, aiding in the identification of vulnerable plaques. [17]
Focal calcification in atherosclerotic plaques is common and increases with age. Although calcification is a good marker for plaque burden, absolute calcium scores do not indicate plaques that are unstable or prone to clinical events but are predictors of future events. An autopsy study has demonstrated a good correlation between plaque size and morphometric analysis of calcification, but no correlation between residual lumen and calcification was identified. [18]
A study comparing the 10-year Framingham risk index, histologic coronary calcification, and culprit plaque morphology in 79 consecutive adults with sudden coronary death demonstrated a modest relationship between the Framingham risk index and the extent of histologic coronary calcification (r = 0.35, P = 0.002). [19] The addition of coronary artery calcium (CAC) score of more than 400 (as assessed by CT scanning) to the Framingham Risk Score has resulted in a higher reclassification rate in the intermediate-risk cohort, thus showing the benefit of imaging. [20]
Cardiovascular disease risk assessment highlights the role of CAC scoring as a gold standard for assessing subclinical atherosclerosis, which can help with risk stratification strategies and estimate the need for lipid-lowering therapy. [21]
Gross Findings
In the aorta, atherosclerotic lesions have been classified largely on gross findings. Fatty streaks are yellow, minimally raised lesions that demonstrate abundant lipid when stained with oil red O. They represent early stages of atherosclerosis. [1] Fibrous plaques are raised, white, firmer areas that are relatively well demarcated and contain a fibrous cap overlying a lipid core. [1] Ulcerated plaques demonstrate surface thrombosis and represent ruptured fibroatheromas, which can lead to acute vascular events. [1]
Coronary lesions, when cut on cross-section, show bright yellow cores when there is abundant extracellular lipid, as in fibroatheromas. Ruptured or eroded plaques demonstrate a luminal thrombus, which is pale red or tan in the unfixed state, depending on the proportion of fibrin, platelets, and entrapped red blood cells. Calcified plaques are hard and brittle, are difficult to cut with a scalpel blade, and must be decalcified during or after fixation so that sections for microscopy may be performed.
The gross findings of carotid plaques are similar to those of the coronary arteries. There is often calcification, which can be seen and felt as mineral deposits. Atheromas are bright yellow on cross-section, and atheromas with intraplaque hemorrhage show a more variegated yellow-red cut surface. Fibrous plaques are homogeneous, firm, and white and often show areas of calcification.
Studies highlight the role of genetic factors, such as the NEDD4L gene, in modulating vascular inflammation and atherosclerosis plaque stability. Genetic polymorphisms, such as the PCSK9 (rs2149041) polymorphism, are associated with increased carotid intima-media thickness (CIMT), which is a marker of early atherosclerosis. [22, 23]
Adaptive Intimal Thickening and Intimal Xanthomas
A study of coronary arteries of 63 hearts obtained from deceased fetuses, infants, children, and adolescents found that coronary intimal thickening begins in fetuses and progresses to atherosclerosis in the pediatric population and adolescents. [24] The most common sites of intimal thickening were near bifurcation sites in the left anterior descending coronary artery (55.6%) and in areas free of bifurcation in the right coronary artery (75%). The extent of intimal thickening was significantly associated with older ages in this population. [24] Optical frequency domain imaging is being explored to assess coronary atherosclerotic plaques. This technology can help in the precise characterization of different types of atherosclerotic lesions, including adaptive intimal thickening and intimal xanthomas. [25]
The early lesions consist of two nonatherosclerotic intimal lesions referred to as adaptive intimal thickening and intimal xanthoma ("fatty streak" in the AHA classification) (see the image below). Intimal xanthoma denotes a lesion rich in foamy macrophages without extracellular lipid pools. Adaptive intimal thickening is present from birth and grows in areas of low shear stress, and it consists mainly of smooth muscle cells in a proteoglycan-rich matrix. Studies suggest that intimal xanthomas may regress and do not always progress to more severe forms of atherosclerosis. [26]
Observations from experimental models and autopsy studies in young human subjects suggest that monocyte adherence to the endothelial surface and transmigration into the intima occur as the earliest events in the development of atherosclerotic lesions. Adaptive intimal thickening is characterized by the retention of modified lipoproteins within the proteoglycan-rich matrix in the intima. The initiation of adhesion increases the expression of selectins, which facilitates the rolling of monocytes, followed by firm attachment by endothelial integrins. Low-density lipoprotein (LDL) oxidation, a critical step in atherosclerosis development, has been shown to occur through the induction of lipoxoygenases, myeloperoxidases, inducible nitric oxide synthase, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (see the image below). Oxidized LDL (ox-LDL) induces endothelial dysfunction, leading to the expression of adhesion molecules such as vascular cell adhesion molecule-1 (i.e., VCAM-1) and E-selectin. These molecules facilitate the adhesion and transmigration of monocytes into the intima, where they differentiate into macrophages and internalize ox-LDL via scavenger receptors, including LOX-1. This process contributes to foam cell formation and the development of intimal xanthomas. [27, 28]
Atherosclerosis pathology. Spectrum of representative coronary lesion morphologies seen in our sudden death population, forming the basis for our modified American Heart Association (AHA) descriptive classification. The two nonprogressive lesions are intimal thickening and intimal xanthomas (foam cell collections known as fatty streaks, AHA type II). Pathologic intimal thickening (PIT) (AHA type III transitional lesions) marks the first of the progressive plaques, as they are the assumed precursors to more advanced fibroatheroma (FA). Thin-cap fibroatheromas (TCFAs) are considered precursors to plaque rupture. Essentially missing from the AHA consensus classification are alternative entities that give rise to coronary thrombosis, namely erosion and the calcified nodule. Erosions can occur on a substrate of PIT or FA, whereas calcified nodules depict eruptive fragments of calcium that protrude into the lumen, causing a thrombotic event. Lastly, healed plaque ruptures are lesions with generally smaller necrotic cores and focal areas of calcification where the surface generally shows areas of healing rich in proteoglycans. Multiple healed plaque ruptures are thought responsible for progressive luminal narrowing. Ca2+ = calcium; EL = extracellular lipid; FC = fibrous cap; NC = necrotic core; Th = luminal thrombus. (Reproduced with permission from Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. May 2000;20(5):1262-75.)
Studies have emphasized the role of vascular smooth muscle cells in atherosclerosis, particularly in plaque stability and progression. Additionally, the presence of fibrin within necrotic cores of advanced plaques suggests its role in intraplaque hemorrhage and plaque instability. [29, 30]
Pathologic Intimal Thickening
PIT or type III lesions in the AHA classification are thought to represent the earliest of the progressive plaques. This lesion is primarily composed of smooth muscle cells near the lumen and matrix consisting chiefly of proteoglycan and type III collagen. Focal areas of accumulated lipid ("lipid pools") are found localized toward the abluminal medial wall as areas devoid of smooth muscle cells but rich in proteoglycans.
It is thought that lesions of PIT that show the presence of macrophages are at a more advanced stage, as demonstrated by Nakashima et al. in their study of early coronary lesions progression near branch points. [31] This study demonstrated that PIT and intimal xanthoma occur simultaneously and are not truly separable lesions. A variable number of T lymphocytes are also observed at this stage, but a true necrotic core is absent. Areas of lipid pools may also contain free cholesterol appearing as cholesterol clefts on paraffin-stained sections.
The formation of lipid pools is associated with the infiltration and deposition of plasma-derived lipids. Intimal smooth muscle cells are dispersed in association with macrophage infiltration and lipid pool formation, without significant proliferation or apoptosis. [32] Studies suggest that the loss of smooth muscle cells (death by apoptosis) may be involved as their remnant basement membranes can be visualized by periodic acid–Schiff (PAS) staining and show microcalcification. In addition, the confirmation of lipids by oil red O staining is highly suggestive of a lipid retention process facilitated by select proteoglycans and oxidation, which may lead to activation of factors responsible for apoptosis.
Progression to Complex Atherosclerotic Lesions
The following generally describes the progression of atherosclerotic lesions (also, see the image below).
-
Fibroatheroma
-
Thin-cap fibroatheroma ("vulnerable plaque") and plaque rupture
-
Necrotic core expansion and risk for plaque rupture
-
Intraplaque hemorrhage and necrotic core expansion
-
Healed plaque rupture
-
Erosion
Atherosclerosis pathology. Simplified scheme for classifying atherosclerotic lesions modified from the current American Heart Association (AHA) recommendations. The boxed areas represent the seven categories of lesions. Dashed lines are used for two categories, because there is controversy over the role that each plays in the initial phase of lesion formation, and both "lesions" can exist without progressing to a fibrous cap atheroma (ie, AHA type IV lesion). The processes responsible for progression are listed between categories. Lines (solid and dotted; dotted lines represent the least-established processes) depict current concepts of how one category may progress to another, with the thickness of the line representing the strength of supportive evidence that the events occur. (Reproduced with permission from Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. May 2000;20(5):1262-75.)
Fibroatheroma
The fibrous cap atheroma is the first of the advanced lesions of coronary atherosclerosis by the AHA classification scheme. Its defining feature is the presence of a lipid-rich necrotic core encapsulated by collagen-rich fibrous tissue. The fibrous cap atheroma may result in significant luminal narrowing and is also prone to complications of surface disruption, thrombosis, and calcification. The origin and development of the core are fundamental to understanding the progression of coronary artery disease. The fibrous cap consists of collagen, smooth muscle cells, and proteoglycan with varying degrees of inflammatory cells — mostly macrophages and lymphocytes. The thickness of the fibrous cap distinguishes the fibroatheroma (relatively thick) from the thin fibrous cap atheroma (classic "vulnerable" plaque). Studies have highlighted the dynamic nature of the fibrous cap, which undergoes continuous remodeling influenced by inflammatory mediators. [33]
Recognition of early necrosis is identified by macrophage infiltration within lipid pools associated with a substantial increase in free cholesterol and breakdown of extracellular matrix, presumably by matrix metalloproteinase (MMP) activity. This, together with the death of macrophages in the setting of defective phagocytic clearance of apoptotic cells, is thought to contribute to the development of late plaque necrosis. Ultimately, the size of the necrotic core is a strong predictor of lesion vulnerability. [31, 34]
Inflammation plays a decisive role in the pathophysiology of atherosclerosis. Proinflammatory cytokines, such as gamma interferon (IFN-γ), can inhibit the synthesis of interstitial collagen by smooth muscle cells, thereby weakening the fibrous cap. Additionally, these cytokines induce enzymes capable of breaking down the arterial extracellular matrix, further destabilizing the plaque. [33] Oxidative stress contributes to the progression of atherosclerosis by promoting endothelial dysfunction and inflammation. [35]
Thin cap fibroatheroma ("vulnerable plaque") and plaque rupture
The thin-cap atheroma is thought to be a precursor lesion to plaque rupture and is characterized by a necrotic core overlaid by a thin, fibrous cap (65 µm or less), which is heavily infiltrated by macrophages and T lymphocytes. The fibrous cap is composed of type I collagen with varying degrees of macrophages and lymphocytes and very few, if any, alpha-actin positive smooth muscle cells.
The density of macrophages at the site of rupture is typically very high, although in some cases macrophages may be sparse. However, because plaque rupture is responsible for 76% of fatal coronary events associated with thrombi in sudden coronary death patients, identification of the thin-cap atheroma is critical. A common mechanism of disruption of the fibrous cap atheroma occurs via the thinning or weakening of the fibrous cap, resulting in fissures and ruptures.
Plaque rupture is defined as an area of fibrous cap disruption in which the overlying thrombus is in contact with the underlying necrotic core.
The luminal thrombus is platelet-rich at the rupture site. Plaque ruptures are most prevalent in the proximal coronary artery near branch points and are frequently found in the proximal left anterior descending coronary artery, followed by the right and left circumflex coronary arteries.
The causes of plaque rupture are poorly understood, but responsible factors include expression of factors that weaken the fibrous cap, such as MMPs, enzymes (e.g., myeloperoxidases) produced by inflammatory cells, high shear regions, stress points, macrophage calcification, and iron deposition. Data are also beginning to emerge that demonstrate critical differences in gene expression between stable and unstable atherosclerotic plaques. [36] In one of these studies, differential expression of 18 genes associated with lesion instability included the metalloproteinase ADAMDEC1, retinoic acid receptor responser-1, cysteine protease legumain (a potential activator of MMPs), and cathepsins. [36]
More recently, in an autopsy study, investigators reported an association between B lymphocytes and macrophages in the perivascular adipose tissue (PvAT) with coronary atherosclerosis. [37] They concluded that the "density of CD20+ B lymphocytes and CD68+ macrophages in periplaque PvAT was increased with plaque size, and the CD68+ macrophages were greater near unstable atherosclerotic plaques than near stable lesions." Moreover, there was more intense inflammation in the periplaque PvAT than in the PvAT distal to the atherosclerotic plaques. [37]
Necrotic core expansion and risk for plaque rupture
Expansion of the necrotic core is an important pathogenic process contributing to plaque vulnerability. The presenting inflammatory stimuli for macrophage recruitment into lipid pools are poorly understood along with the respective signaling pathways for subsequent apoptotic cell death and necrosis. Evidence exists pointing toward the involvement of the endoplasmic reticulum stress pathway, or so-called unfolded protein response, as the primary mechanism of macrophage cell death in plaques. This pathway promotes the death of macrophages — the resultant accumulation of dead macrophages coupled with defective phagocytic clearance has been cited as one of the principal factors causing necrotic core expansion.
Intraplaque hemorrhage and necrotic core expansion
Intraplaque hemorrhage plays a pivotal role in the progression and destabilization of atherosclerotic plaques, significantly contributing to necrotic core expansion as red blood cells are a rich source of free cholesterol, which is an important constituent of ruptured plaques. [38, 39] The red blood cells are enriched with lipids constituting 40% of their weight and free cholesterol content within membranes exceeding all other cell types. The expression of glycophorin-A (a protein exclusive to red blood cell membranes) within the necrotic cores of advanced coronary atheroma is strongly positive, whereas its presence in plaques with early necrosis or PIT remains absent or low.
Intraplaque hemorrhage likely occurs from leaky vasa vasorum that infiltrate the plaque as the lesion thickness increases. The authors have reported that microvessel density is increased in advanced plaques compared with early plaques. Microvessels in normal and atherosclerotic arteries are thin-walled, with compromised structural integrity characterized by poor endothelial junctions. [40] Therefore, intraplaque hemorrhage together with the death of macrophages in the setting of defective phagocytic clearance of apoptotic cells is thought to contribute to the development of necrotic core in advanced plaques.
Healed plaque rupture
Plaque rupture can lead to either thrombosis or healing, depending on various factors including inflammatory responses and local shear stress. The healing process involves thrombus lysis, granulation tissue formation, and reendothelialization, promoting plaque repair and vessel integrity. [41]
Morphologic studies suggest that plaque progression beyond 40-50% cross-sectional luminal narrowing occurs secondary to repeated ruptures. Ruptured lesions with healed repair sites, namely, healed plaque ruptures (HPRs) as shown by Mann and Davies, are easily detected microscopically by the identification of breaks in the fibrous cap with an overlying repair reaction consisting of proteoglycans and/or collagen, depending on the phase of healing. [42] Early-healed lesions are rich in proteoglycans, which are eventually replaced by type I collagen.
HPRs play a significant role in the progression of atherosclerotic lesions, contributing to luminal narrowing and increased plaque burden. Histopathological analyses have demonstrated that repeated cycles of rupture and healing can lead to significant stenosis. For instance, Burke et al. found that healed ruptures were present in 61% of cases of sudden coronary death, with an even higher prevalence in cases with stable plaque (80%). [43]
A meta-analysis encompassing 13 studies with 3141 patients revealed that the overall incidence of healed plaques was 40% (95% CI: 39-42). Notably, the incidence was higher in patients with stable angina pectoris (46%) than in those with ACS (37%). Additionally, healed plaques were more prevalent in culprit lesions (48%) than in non-culprit lesions (24%). This underscores the significance of HPRs in both stable and unstable coronary artery disease. [44]
The prevalence of silent ruptures in the clinical population is unknown. Few angiographic studies have demonstrated plaque progression, and short-term studies have suggested that thrombosis is the likely cause. Mann and Davies showed that the frequency of HPR increases along with lumen narrowing. [42] In plaques with 0-20% diameter stenosis, the incidence of HPRs was 16%; in lesions with 21-50% stenosis, the incidence was 19%; and in plaques with >50% narrowing, the incidence was 73%. [42]
The authors have shown a high frequency of HPRs in the coronary arteries in patients dying suddenly with severe coronary disease. [45] The percentage of cross-sectional luminal narrowing was dependent on the number of healed repair sites.
Studies have shown that certain conditions can influence plaque stability and healing. For instance, diabetes mellitus is associated with a higher incidence of HPRs and increased arterial calcification, indicating a more active atherogenic process. [46]
The presence of HPRs is associated with a higher incidence of thin-cap FAs, plaque rupture, microvessel formation, macrophage accumulation, and calcification. These features are indicative of plaque vulnerability and highlight the importance of monitoring patients with healed plaques for potential adverse cardiovascular events. [44]
Erosion
Although plaque rupture is the most common cause of coronary thrombosis, acute coronary syndromes may occur in the absence of rupture. As mentioned earlier, thrombi may occur as a result of three different events: plaque rupture, plaque erosion, or, rarely, a calcified nodule (see Etiology). Plaque erosion is characterized by the absence of the endothelium at the site of erosion, with exposed intima composed of smooth muscle cells and proteoglycans, as well as typically minimal inflammation. Studies have shown that eroded surfaces contain fewer macrophages and T lymphocytes compared to ruptured plaques, indicating a different cellular composition and inflammatory profile. [47]
Studies have identified clinical and laboratory predictors for plaque erosion in patients with ACS. These predictors include age less than 68 years, anterior ischemia, absence of diabetes mellitus, hemoglobin levels greater than 15.0 g/dL, and normal renal function. When all these parameters are present in a patient with non–ST‐segment elevation‐ACS (i.e., NSTE-ACS), the probability of plaque erosion increases significantly. [48]
In a series of 20 patients who died with acute MI, van der Wal et al found plaque ruptures in 60% of lesions with thrombi, whereas the remaining 40% showed "superficial erosion." [49] The term "erosion" was chosen because the luminal surface beneath the thrombus lacked endothelial cells. In these lesions, the thrombus was confined to the most luminal portion of the plaque, and there was an absence of ruptures following serial sectioning of these lesions.
In addition, the authors' laboratory studied nearly 100 cases of sudden coronary death and found that 60% of all thrombi could be attributed to plaque rupture and 40% to erosions. Morphologically, major differences exist in the cellular composition of ruptured vs erosion lesions. Unlike the prominent fibrous cap inflammation described in ruptures, eroded surfaces contain few macrophages (rupture 100% vs erosion 50%, P < 0.0001) and T lymphocytes (rupture 75% vs erosion 32%, P < 0.004). Cell activation, indicated by human lymphocyte antigen (HLA)-DR staining, was identified in macrophages and T cells in 89% of plaque ruptures and in 36% of plaque erosions (P = 0.0002). [50] The smooth muscle cells near the erosion site appeared "activated," often displaying bizarre shapes with hyperchromatic nuclei and prominent nucleoli. The incidence of calcification was also less common in erosion than in ruptures.
The authors have also shown that more than 85% of thrombi in erosions exhibited late stages of healing characterized by acute inflammatory cell lysis, invasion by smooth muscle cells and/or endothelial cells, or organized layers of smooth muscle cells and proteoglycans with varying degrees of platelet/fibrin layering, whereas in ruptures only one half of thrombi showed healing. [51] Postmortem coronary thrombi superimposed on eroded plaques have been shown to contain a higher density of myeloperoxidase-positive cells than those superimposed on ruptured plaques. [52] Also, circulation blood myeloperoxidase levels are elevated in patients with acute coronary sinus with erosion compared with those with rupture, suggesting that elevations in selective inflammatory biomarkers may reflect specific acute coronary events.
Probe electrospray ionization mass spectrometry appears to have the potential to detect new biomarkers of atherosclerosis (e.g., cholesterol sulfate, phospholipid PE18:0/24:0), although current findings are from animal models (rabbits). [53]
Comparison of plaque rupture and plaque erosion
Both clinical and morphologic differences are widely apparent between plaque rupture and erosion. Beginning with age, patients with plaque rupture tend to be significantly older (53 ± 10 y) than those with erosion (44 ± 7 y) (P < 0.02). Survival is also a critical factor because an estimation of fatal ruptures in the fifth decade of life is 17 per 100,000 per year compared with 6 per 100,000 for plaque erosion.
Although the relationship between risk factors and culprit plaques is similar between females and males, the proportion of females younger than the age of 50 years dying suddenly with plaque erosion is remarkably higher. Plaque burden expressed as the percentage of cross-sectional area stenosis excluding the thrombus is greater in plaque ruptures (78 ± 12%) than erosions (70 ± 11%) (P < 0.03), whereas eccentric plaques are more common in erosions. Unlike the prominent fibrous cap inflammation described in ruptures, eroded surfaces contain fewer macrophages and T lymphocytes. Taken together, eroded plaques tend to be eccentric lesions rich in smooth muscle cells and proteoglycans with very little inflammation or calcification.
Immunohistochemistry
Immunohistochemistry is not necessary for the diagnosis or classification of atherosclerotic plaques. However, immunolocalization of both cellular and acellular elements within human and animal lesions has contributed greatly to the understanding of atheroslcerotic plaque progression. Immunohistochemical stains for smooth muscle cell antigens (e.g., smooth muscle actin), inflammatory cells (especially macrophages and T lymphocytes), blood-derived antigens (e.g., fibrin and platelet surface antigens), and enzymes (e.g., elastases and collagenases) have all been used to characterize the components of atherosclerotic plaques, and these represent only a sample.
Immunoreactivity to CD31 is a crucial marker in diagnostic pathology. CD31 is a protein expressed on endothelial cells, which plays a role in leukocyte transmigration and angiogenesis within plaques. [54, 55]
-
Atherosclerosis pathology. Spectrum of representative coronary lesion morphologies seen in our sudden death population, forming the basis for our modified American Heart Association (AHA) descriptive classification. The two nonprogressive lesions are intimal thickening and intimal xanthomas (foam cell collections known as fatty streaks, AHA type II). Pathologic intimal thickening (PIT) (AHA type III transitional lesions) marks the first of the progressive plaques, as they are the assumed precursors to more advanced fibroatheroma (FA). Thin-cap fibroatheromas (TCFAs) are considered precursors to plaque rupture. Essentially missing from the AHA consensus classification are alternative entities that give rise to coronary thrombosis, namely erosion and the calcified nodule. Erosions can occur on a substrate of PIT or FA, whereas calcified nodules depict eruptive fragments of calcium that protrude into the lumen, causing a thrombotic event. Lastly, healed plaque ruptures are lesions with generally smaller necrotic cores and focal areas of calcification where the surface generally shows areas of healing rich in proteoglycans. Multiple healed plaque ruptures are thought responsible for progressive luminal narrowing. Ca2+ = calcium; EL = extracellular lipid; FC = fibrous cap; NC = necrotic core; Th = luminal thrombus. (Reproduced with permission from Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. May 2000;20(5):1262-75.)
-
Atherosclerosis pathology. Simplified scheme for classifying atherosclerotic lesions modified from the current American Heart Association (AHA) recommendations. The boxed areas represent the seven categories of lesions. Dashed lines are used for two categories, because there is controversy over the role that each plays in the initial phase of lesion formation, and both "lesions" can exist without progressing to a fibrous cap atheroma (ie, AHA type IV lesion). The processes responsible for progression are listed between categories. Lines (solid and dotted; dotted lines represent the least-established processes) depict current concepts of how one category may progress to another, with the thickness of the line representing the strength of supportive evidence that the events occur. (Reproduced with permission from Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. May 2000;20(5):1262-75.)
Tables
|
Description |
Thrombosis |
Nonatherosclerotic intimal lesions |
||
Intimal thickening |
Normal accumulation of smooth muscle cells (SMCs) in the intima in the absence of lipid or macrophage foam cells |
Absent |
Intimal xanthoma |
Superficial accumulation of foam cells without a necrotic core or fibrous cap; based on animal and human data, such lesions usually regress |
Absent |
Progressive atherosclerotic lesions |
||
Pathologic intimal thickening |
SMC-rich plaque with proteoglycan matrix and focal accumulation of extracellular lipid |
Absent |
Fibrous cap atheroma |
Early necrosis: focal macrophage infiltration into areas of lipid pools with an overlying fibrous cap Late necrosis: loss of matrix and extensive cellular debris with an overlying fibrous cap |
Absent |
Thin cap fibroatheroma |
A thin, fibrous cap (< 65 µm) infiltrated by macrophages and lymphocytes with rare or absence of SMCs and a relatively large underlying necrotic core; intraplaque hemorrhage/fibrin may be present |
Absent |
Lesions with acute thrombi |
||
Plaque rupture |
Fibroatheroma with fibrous cap disruption; the luminal thrombus communicates with the underlying necrotic core |
Occlusive or nonocclusive |
Plaque erosion |
Plaque composition, as above; no communication of the thrombus with the necrotic core; can occur on a plaque substrate of pathologic intimal thickening or fibroatheroma |
Usually nonocclusive |
Calcified nodule |
Eruptive (shedding) of calcified nodules with an underlying fibrocalcific plaque with minimal or absence of necrosis |
Usually nonocclusive |
Lesions with healed thrombi |
||
Fibrotic (without calcification) Fibrocalcific (+/− necrotic core) |
Collagen-rich plaque with significant luminal stenosis; lesions may contain large areas of calcification with few inflammatory cells and minimal or absence of necrosis; these lesions may represent healed erosions or ruptures |
Absent |
* Modified from Virmani et al [2] Lesion reference to AHA types V and VI was discarded, because it failed to account for the three different morphologies (rupture, erosion, and calcified nodule) that give rise to acute coronary thrombosis. |
||
