Practice Essentials
Pseudohypoparathyroidism (PHP) is a heterogeneous group of rare endocrine disorders characterized by normal renal function and resistance to the action of parathyroid hormone (PTH), manifesting with hypocalcemia, hyperphosphatemia, and increased serum concentration of PTH. [1] There are five variants of pseudohypoparathyroidism: PHP type 1a (PHP-1a), PHP type 1b (PHP-1b), PHP type 1c (PHP-1c), PHP type 2 (PHP-2), and pseudopseudohypoparathyroidism (PPHP). PHP type 1a is the most common subtype and represents 70% of cases. [2]
In 1942, Fuller Albright first introduced the term pseudohypoparathyroidism to describe patients who presented with PTH-resistant hypocalcemia and hyperphosphatemia along with an unusual constellation of developmental and skeletal defects, collectively termed Albright hereditary osteodystrophy (AHO). These features included short stature, rounded face, shortened fourth metacarpals and other bones of the hands and feet, obesity, dental hypoplasia, and soft-tissue calcifications/ossifications. In addition, administration of PTH failed to produce the expected phosphaturia or to stimulate renal production of cyclic adenosine monophosphate (cAMP). However, the AHO phenotype is not a feature of PHP-1b or PHP-2.
The molecular defects in the gene (GNAS1) encoding the α subunit of the stimulatory G protein (Gsα) contribute to at least four different forms of the disease: PHP-1a, PHP-1b, PHP-1c, and PPHP. [3] While PHP-2 is associated with renal resistance to PTH action, the genetic abnormalities causing PHP-2 remain to be identified. [4]
Diagnosis of PHP is defined by the coexistence of hypocalcemia and hyperphosphatemia with elevated PTH levels in the presence of normal vitamin D values and normal renal function and the absence of hypercalciuria. Pseudohypoparathyroidism can be diagnosed by blood or urine tests to measure the levels of calcium, phosphorous, and parathyroid hormone. Genetic testing for a mutation in the GNAS1 gene can confirm diagnosis and identify subtype. [2]
The goals of pharmacotherapy are to correct calcium deficiency, prevent complications, and reduce morbidity. Intravenous calcium is the initial treatment for all patients with severe symptomatic hypocalcemia. Administration of oral calcium and 1alpha-hydroxylated vitamin D metabolites, such as calcitriol, remains the mainstay of treatment and should be initiated in every patient with a diagnosis of PHP. Maintaining serum total and ionized calcium levels within the reference range discourages hypercalciuria and suppresses PTH levels to normal. Patients with intracranial calcifications may experience seizures related to chronic neuropathic changes, and they may need antiepileptic medications. [5, 6]
(See the image below.)
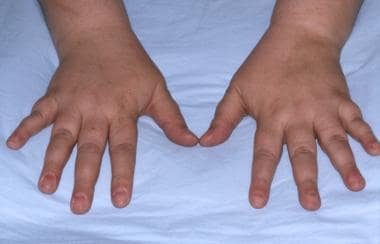 Patient with pseudohypoparathyroidism showing shortened fourth metacarpals.
Patient with pseudohypoparathyroidism showing shortened fourth metacarpals.
Pathophysiology
Genetics
A heterozygous mutation of the GNAS gene that encodes the G stimulatory α subunit (Gsα) of guanine nucleotide-binding protein leads to a loss of expression or function of the Gsα, [7] which impairs the transmission of stimulatory signals to adenylate cyclase, limiting cyclic AMP (cAMP) generation necessary for hormone action. Inactivation of GNAS mutations involving Gsα can involve skeletal anomalies such as short metacarpals and/or metatarsals in PHP-1a and PPHP. [8] Reyes et al reported skeletal abnormalities in an extended Belgian family in the absence of abnormal calcium and phosphate regulation that were attributed to a novel heterozygous splice-site mutation in PTHLH exon 3. [8] The investigators indicated that PTHrP (PTH-related protein) haploinsufficiency results in skeletal anomalies similar to those seen in heterozygous GNAS mutations.
GNAS mutations on maternally inherited alleles (PHP-1a and PHP-1c) manifest resistance to parathyroid hormone (PTH), thyroid-stimulating hormone (TSH), growth-hormone–releasing hormone (GHRH), and gonadotropins, as well as the the phenotypic features of Albright hereditary osteodystrophy (AHO). [4, 9]
GNAS mutations on paternally inherited alleles (PPHP) have only the phenotypic features of AHO without hormonal resistance. [4] Davies et al reported an analysis of pedigrees of families that included patients with PHP and PPHP, suggesting that patients who inherit the defective gene from the father have pseudo-PHP, because the mutant gene is not expressed and the product of a single maternally inherited GNAS1 gene preserves normal responses to PTH and thyrotropin. [10] However, the occurrence of AHO in patients with pseudo-PHP indicates that one GNAS1 gene is not sufficient in all tissues.
Those with PHP-1b lack typical features of AHO but may have mild brachydactyly. Familial PHP-1b displays an isolated loss of methylation at exon A/B associated with a recurrent 3-kb deletion in the STX16 gene. NESP55 and NESPAS deletions have also been described leading to the loss of all maternal GNAS imprints (epimutations). Sporadic PHP-1b is characterized by complete loss of methylation at the NESPas, XLas, and A/B promoters. In some cases, paternal 20q disomies have been found. [3, 11]
Reyes et al reported the case of a patient with PHP-1b who had a novel GNAS duplication linked to loss of methylation restricted to exon A/B [12] ; Kiuchi et al reported an example of transmission ratio distortion, in which preferential maternal transmission of STX16-GNAS mutations were responsible for autosomal dominant PHP-1b [13] ; and Hanna et al reported a novel familial PHP1b variant in which there was incomplete loss of methylation at GNAS A/B and enhanced methylation at GNAS subdomain AS2 but in the absence of an STX16 deletion on the maternal and paternal differentially methylated regions. [14] In contrast, Iwasaki et al revealed that hypomethylation occurred at GNAS AS2 with genetic alterations located centromeric but not telomeric in patients with PHP-1b. [15]
PHP-2 is associated with renal resistance to PTH action and the absence of AHO phenotype; however, the genetic abnormalities causing PHP-2 remain to be identified. [4]
Testotoxicosis
Testotoxicosis with PHP-1a can occur. Gonadotropin-independent sexual precocity has been reported in 2 boys who presented in infancy with classic PHP-1a. Usually, patients with PHP-1a show resistance to luteinizing hormone, which could lead to primary testicular insufficiency. The paradoxical presentation of testotoxicosis in these boys resulted from an identical point mutation in the GNAS1 gene, which caused both a loss and gain of Gsα function.
PHP-1a, characterized by a loss of Gsα function, is caused by thermal inactivation of the mutant protein at body temperature. Testotoxicosis indicates an organ-specific gain of Gsα function, resulting from the expression of the mutant protein. The lower temperature of the testes protects the mutant protein from thermal inactivation.
Growth plate defects
A study by Sanchez et al found that an imprinting defect in GNAS may lead to growth plate defects in patients with PHP-1b, including brachydactyly and Madelung deformity. This suggests that GNAS signaling has a more extensive role in chondrocyte maturation than was previously believed. [16]
Etiology
PHP type 1a
Several other peptide hormones, including thyroid-stimulating hormone (thyrotropin), antidiuretic hormone, gonadotropins, glucagons, adrenocorticotropin, and growth hormone–releasing hormone, use the α subunit of stimulatory G protein to enhance cAMP production. Patients with PHP-1a can present with resistance to the effects of any of these hormones, although in most patients, responses to corticotropin and glucagon are clinically unaffected.
The dominant pattern of inheritance of PHP-1a has been attributed to haploinsufficiency of GNAS1, meaning that the protein produced by a single normal Gsα allele cannot support normal function, although it may suffice for survival. The single normal Gsα allele preserves the responses to hormones such as corticotropin and glucagon. The haploinsufficiency of the GNAS1 gene is tissue specific, which may explain the selective resistance to hormones and the characteristic habitus of patients with PHP-1a.
Pseudo-PHP
Pseudopseudohypoparathyroidism (PPHP) is caused by GNAS mutations on paternally inherited alleles. Paternal inheritance accounts for differences in the same family where some patients with a defective GNAS1 gene inherited maternally have resistance to PTH (PHP-1a), whereas others with PPHP share with them the habitus of AHO but are not resistant to PTH.
PHP type 1b
Familial PHP-1b is caused by heterozygous deletions in STX16, NESP55, and/or AS exon. Sporadic PHP-1b is characterized by complete loss of methylation at the NESPas, XLas, and A/B promoters. In some cases, paternal 20q disomies have been found. [3] The absence of PTH resistance in the mother and maternal grandfather who carried the same mutation was consistent with current models of paternal imprinting of the GNAS1 gene. [17]
PHP type 1c
PHP-1c appears to be a variant of PHP-1a, in which the specific GNAS mutation disrupts receptor-mediated activation of adenylyl cyclase but does not affect receptor-independent activation of the enzyme. This accounts for the inability to demonstrate reduced activity of solubilized Gsα with conventional assays. [4, 11]
Epidemiology
The estimated prevalence of PHP type 1a, type1b, and PPHP is 1 per 150,000 in Italy. [18] In Japan, the estimated prevalence of PHP type 1a and type 1b is 1 per 295,000. [19, 18] PHP occurs approximately twice as frequently in females as in males. Onset of endocrine symptoms occurs during childhood, although cases with severe hypothyroidism at neonatal screening have been reported. [18]
-
Patient with pseudohypoparathyroidism showing shortened fourth metacarpals.



