Practice Essentials
Idiopathic pulmonary fibrosis (IPF) is defined as a specific form of chronic, progressive fibrosing interstitial pneumonia of unknown cause, primarily occurring in older adults, limited to the lungs, and associated with the histopathologic and/or radiologic pattern of usual interstitial pneumonia (UIP). [1] It causes lung scarring, which, over time, results in reduced oxygen intake.
Signs and symptoms
The clinical symptoms of idiopathic pulmonary fibrosis are nonspecific and can be shared with many pulmonary and cardiac diseases. Most patients present with a gradual onset (often >6 mo) of exertional dyspnea and/or a nonproductive cough. Approximately 5% of patients have no presenting symptoms when idiopathic pulmonary fibrosis is serendipitously diagnosed.
Associated systemic symptoms that can occur but are not common in idiopathic pulmonary fibrosis include the following:
-
Weight loss
-
Low-grade fevers
-
Fatigue
-
Arthralgias
-
Myalgias
See Clinical Presentation for more detail.
Diagnosis
It is critical to obtain a complete history, including medication history, drug use, social history, occupational, recreational, and environmental respiratory exposure history, risks for the human immunodeficiency virus, and review of systems, to ensure other causes of interstitial lung disease are excluded. The diagnosis of idiopathic pulmonary fibrosis relies on the clinician to integrate and correlate the clinical, laboratory, radiologic, and/or pathologic data. [2]
The diagnosis of IPF requires the following [3] :
-
Exclusion of known causes of interstitial lung disease
-
Presence of the high-resolution computed tomography (HRCT) pattern of Typical or Probable UIP
-
Specific combinations of HRCT patterns and histopathology patterns in patients subjected to lung tissue sampling
-
Multi-Displinary discussion between Pulmonary, Radiology and Pathology specialists.
Physical examination in patients with idiopathic pulmonary fibrosis may reveal the following:
-
Fine bibasilar inspiratory crackles (Velcro crackles): Noted in most patients
-
Digital clubbing (25-50%)
-
Pulmonary hypertension at rest (20-40%) [4] : Loud P2 component of the second heart sound, a fixed split S2, a holosystolic tricuspid regurgitation murmur, pedal edema
Laboratory testing
Results from routine laboratory studies are nonspecific for the diagnosis of idiopathic pulmonary fibrosis. Some tests that may be helpful to exclude other causes of interstitial lung disease include the following:
-
Antinuclear antibodies or rheumatoid factor titers: Positive results in about 30% of patients with IPF, but the titers are generally not high [5] . The presence of high titers may suggest a connective tissue disease
-
C-reactive protein level and erythrocyte sedimentation rate: Elevated but nondiagnostic in idiopathic pulmonary fibrosis
-
Complete blood cell count: polycythemia (rare)
-
Arterial blood gas analysis: chronic hypoxemia (common)
Physiologic Assessment:
-
Pulmonary function studies: Nonspecific findings of a restrictive ventilatory defect and reduced diffusion capacity for carbon monoxide (DLCO) [6]
-
6-minute walk test (6MWT) is often used in the initial and longitudinal clinical assessment of patients with idiopathic pulmonary fibrosis. In patients who desaturate to less than 88% during a 6MWT, a progressive decline in the DLCO (>15% after 6 mo) is a strong predictor of increased mortality. [7]
Imaging studies
-
High-resolution computed tomography (HRCT) scanning: Sensitive, specific, and essential for the diagnosis of idiopathic pulmonary fibrosis. Demonstrates patchy, peripheral, subpleural, and bibasilar reticular opacities. Needs HRCT images from clinic *******
-
Chest radiography: Abnormal findings but lacks diagnostic specificity. Demonstrate peripheral reticular opacities (netlike linear and curvilinear densities) predominantly at the lung bases, honeycombing (coarse reticular pattern), and lower lobe volume loss [8] (see the image below)
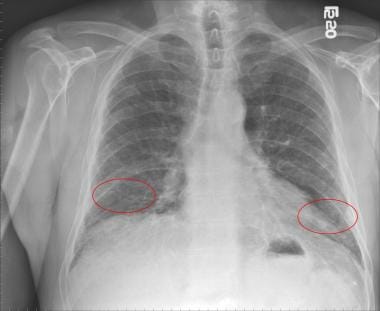 Chest radiograph of a patient with idiopathic pulmonary fibrosis showing bilateral lower lobe reticular opacities (red circles).
Chest radiograph of a patient with idiopathic pulmonary fibrosis showing bilateral lower lobe reticular opacities (red circles).
-
Transthoracic echocardiography: Detects pulmonary hypertension well but has variable performance in patients with idiopathic pulmonary hypertension and other chronic lung disease [4]

Procedures
-
Bronchoscopy: Absence of lymphocytosis in bronchoalveolar lavage fluid may be important for the diagnosis (increased neutrophils [70-90% of patients] and eosinophils [40-60% of all patients]). This procedure may be used to exclude alternative diagnoses.
-
Transbronchial lung cryobiopsy (TBLC) is an acceptable alternative to surgical lung biopsy (SLB) for making a histopathological diagnosis in patients with ILD of undetermined type in medical centers with experience performing TBLC; evidence from latest ATS/ERS guidelines 2022.
-
Surgical lung biopsy (via open lung biopsy or video-assisted thoracoscopic surgery [VATS] [preferred]): Best sample for distinguishing Typical Pathologic usual interstitial pneumonia pattern from other idiopathic interstitial pneumonias and other alternative diagnoses.
See Workup for more detail.
Management
The optimal medical therapy for the treatment of idiopathic pulmonary fibrosis has yet to be identified. Treatment strategies for idiopathic pulmonary fibrosis include the assessment and management of comorbid conditions according to current practice guidelines, including chronic obstructive pulmonary disease, obstructive sleep apnea, gastroesophageal reflux disease, and coronary artery disease.
Other management strategies include the following:
-
Encourage tobacco users to quit and offer pharmacotherapy as needed.
-
Prescribe oxygen therapy in patients with hypoxemia at rest or with exercise (partial pressure of oxygen [PaO2] < 55 mmHg or an oxygen saturation by pulse oximetry [SpO2] < 88%). The goal is to maintain an oxygen saturation of at least 90% at rest, with sleep, and with exertion.
-
Vaccinate patients against influenza and pneumococcal infection.
Surgery
-
Lung transplantation: Refer all patients with diagnosed or probable idiopathic pulmonary fibrosis for lung transplantation evaluation regardless of the vital capacity, unless there are contraindications. [9]
Pharmacotherapy
-
Tyrosine kinase inhibitors (eg, nintedanib)
-
Antifibrotic agents (eg, pirfenidone)
-
Treatment of idiopathic pulmonary fibrosis exacerbation
-
potential ongoing research
See Treatment and Medication for more detail.
Background
Idiopathic pulmonary fibrosis (IPF) is defined as a specific form of chronic, progressive fibrosing interstitial pneumonia of unknown cause, primarily occurring in older adults, limited to the lungs, and associated with the histopathologic and/or radiologic pattern of usual interstitial pneumonia (UIP). [1]
Of the seven listed idiopathic interstitial pneumonias in the American Thoracic Society/European Respiratory Society consensus statement (ie, idiopathic pulmonary fibrosis, nonspecific interstitial pneumonia, cryptogenic organizing pneumonia, acute interstitial pneumonia, desquamative interstitial pneumonia, respiratory bronchiolitis-associated interstitial pneumonia, lymphoid interstitial pneumonia), idiopathic pulmonary fibrosis is the most common. [10] Idiopathic pulmonary fibrosis portends a poor prognosis, and, to date, no proven effective therapies are available for the treatment of idiopathic pulmonary fibrosis beyond lung transplantation. [2]
Most patients with idiopathic pulmonary fibrosis present with a gradual onset, often greater than six months, of dyspnea and/or a nonproductive cough. The symptoms often precede the diagnosis by a median of one to two years. [11] A chest radiograph typically reveals diffuse reticular opacities. However, it lacks diagnostic specificity. [12] High-resolution computed tomography (HRCT) findings are significantly more sensitive and specific for the diagnosis of idiopathic pulmonary fibrosis. On HRCT images, usual interstitial pneumonia is characterized by the presence of reticular opacities often associated with traction bronchiectasis. As idiopathic pulmonary fibrosis progresses, honeycombing becomes more prominent. [8] Pulmonary function tests often reveal restrictive impairment and reduced diffusing capacity for carbon monoxide. [12]
Available data suggest that no single etiologic agent serves as a common inciting event in the pathogenesis of idiopathic pulmonary fibrosis. During the past 15 years, the pathogenesis theory of generalized inflammation progressing to widespread parenchymal fibrosis has become less popular. [12] Rather, it is now believed that epithelial injury and activation in fibroblast foci are crucial early events that trigger a cascade of changes leading to reorganization of pulmonary tissue compartments. [13]
As mentioned above, idiopathic pulmonary fibrosis is an idiopathic interstitial pneumonitis characterized by usual interstitial pneumonia on histopathology. The hallmark pathologic feature of usual interstitial pneumonia is a heterogeneous, subpleural, variegated appearance with alternating areas of healthy lung, interstitial inflammation, fibroblastic foci formation and honeycomb change. Fibrosis predominates over inflammation. [13]
The diagnosis of idiopathic pulmonary fibrosis relies on the multi-discplinary integration of the clinical, laboratory, radiologic and/or pathologic data to make a clinical-radiologic-pathologic correlation that supports the diagnosis of idiopathic pulmonary fibrosis. [2]
Pathophysiology
The previous theory regarding the pathogenesis of idiopathic pulmonary fibrosis (IPF) was that generalized inflammation progressed to widespread parenchymal fibrosis. However, anti-inflammatory agents and immune modulators have proved to be minimally effective in modifying the natural course of the disease. It is currently believed that idiopathic pulmonary fibrosis is an epithelial-fibroblastic disease, in which unknown endogenous or environmental stimuli disrupt the homeostasis of alveolar epithelial cells, resulting in diffuse epithelial cell activation and aberrant epithelial cell repair. [14]
In the current hypothesis regarding the pathogenesis of idiopathic pulmonary fibrosis, exposure to an inciting agent (eg, smoke, environmental pollutants, environmental dust, viral infections, gastroesophageal reflux disease, chronic aspiration) in a susceptible host may lead to the initial alveolar epithelial damage. [15] Reestablishing an intact epithelium following injury is a key component of normal wound healing. In idiopathic pulmonary fibrosis, it is believed that after injury, aberrant activation of alveolar epithelial cells provokes the migration, proliferation, and activation of mesenchymal cells with the formation of fibroblastic/myofibroblastic foci, leading to the exaggerated accumulation of extracellular matrix with the irreversible destruction of the lung parenchyma. [15]
Activated alveolar epithelial cells release potent fibrogenic cytokines and growth factors. These include, tumor necrosis factor-α (TNF-α), transforming growth factor-β (TGF-β), platelet-derived growth factor, insulin-like growth factor-1, and endothelin-1 (ET-1). [13, 15, 16] These cytokines and growth factors are involved in the migration and proliferation of fibroblasts and the transformation of fibroblasts into myofibroblasts. Fibroblasts and myofibroblasts are key effector cells in fibrogenesis, and myofibroblasts secrete extracellular matrix proteins. [15, 17]
For normal wound healing to occur, wound myofibroblasts must undergo apoptosis. Failure of apoptosis leads to myofibroblast accumulation, exuberant extracellular matrix protein production, persistent tissue contraction, and pathologic scar formation. [15] TGF-β has been shown to promote an antiapoptotic phenotype in fibroblasts. [15] Additionally, myofibroblasts in fibroblastic foci of idiopathic pulmonary fibrosis have been reported to undergo less apoptotic activity in comparison to myofibroblasts in the fibromyxoid lesions of bronchiolitis obliterans organizing pneumonia. [18]
Excess alveolar epithelial cell apoptosis and fibroblast resistance to apoptosis are also believed to contribute to fibroproliferation in idiopathic pulmonary fibrosis. Research has demonstrated that prostaglandin E2 deficiency, in lung tissue of patients with pulmonary fibrosis, results in increased sensitivity of alveolar epithelial cells to FAS-ligand induced apoptosis but induces fibroblast resistance to Fas-ligand induced apoptosis. [19] Therefore, apoptosis resistance in the fibroblasts and myofibroblasts participating in the repair of the alveolar epithelium may contribute to the persistent and/or progressive fibrosis in idiopathic pulmonary fibrosis.
Evidence for a genetic basis for idiopathic pulmonary fibrosis is accumulating. It is estimated that 10-20 % of IPF meet the criteria for familial disease. So far there has been two types of genetic variants associated with the increased risk of this disease. There are the common (MUC5b, TOLLIP, etc...) and the rare (Surfactant and telomere maintenance pathway) variants.
It has been described that mutant telomerase is associated with familial idiopathic pulmonary fibrosis. [20] Telomerase is a specialized polymerase that adds telomere repeats to the ends of chromosomes. This helps to offset shortening that occurs during DNA replication. TGF-β negatively regulates telomerase activity. [15] It is proposed that pulmonary fibrosis in patients with short telomeres is provoked by a loss of alveolar epithelial cells. Telomere shortening also occurs with aging, and it can also be acquired. This telomere shortening could promote the loss of alveolar epithelial cells, resulting in aberrant epithelial cell repair, and therefore should be considered as another potential contributor to the pathogenesis of idiopathic pulmonary fibrosis. [20]
Additionally, a common variant in the putative promoter of the gene that encodes mucin 5B (MUC5B) has been associated with the development of both familial interstitial pneumonia and sporadic pulmonary fibrosis. MUC5B expression in the lung was reported to be 14.1 times as high in subjects who had idiopathic pulmonary fibrosis as in those who did not. Therefore, dysregulated MUC5B expression in the lung may be involved in the pathogenesis of pulmonary fibrosis. [21]
Finally, caveolin-1 has been proposed as a protective regulator of pulmonary fibrosis. Caveolin-1 limits TGF-β–induced production of extracellular matrix proteins and restores the alveolar epithelial-repair process. [15] It has been observed that the expression of caveolin-1 is reduced in lung tissue from patients with idiopathic pulmonary fibrosis and that fibroblasts, the key cellular component of fibrosis, have low levels of caveolin-1 expression in patients with idiopathic pulmonary fibrosis. [22]
The recognition of the above-mentioned factors as contributors to the pathogenesis of idiopathic pulmonary fibrosis has led to the development of novel approaches to treat idiopathic pulmonary fibrosis.
Etiology of IPF
The etiology of idiopathic pulmonary fibrosis (IPF) remains undefined; however, in the current hypothesis regarding the pathogenesis of idiopathic pulmonary fibrosis, exposure to an inciting agent (eg, smoke, environmental pollutants, environmental dust, viral infections, gastroesophageal reflux disease, chronic aspiration) in a susceptible host may lead to the initial alveolar epithelial damage. [15] This damage may lead to activation of the alveolar epithelial cells, which provokes the migration, proliferation, and activation of mesenchymal cells with the formation of fibroblastic/myofibroblastic foci, leading to the exaggerated accumulation of extracellular matrix with the irreversible destruction of the lung parenchyma. [15]
The following is a summary of possible inciting factors:
-
Occupational dust or fume exposure (eg, hairdressers, ranchers, farmers, stonecutters, metal workers)
-
Older age (approximately two thirds of patients are >60 years at diagnosis)
-
Smoking history
-
Male sex (higher prevalence in men vs women)
-
Genetics
Other potential causes of idiopathic pulmonary fibrosis have been recognized through the study of familial pulmonary fibrosis. Familial pulmonary fibrosis, affecting two or more members of the same primary biological family, accounts for 10-20% of total patients with idiopathic pulmonary fibrosis. [23]
Genetic mutations in serum surfactant protein C have been discovered in some individuals with familial pulmonary fibrosis. [23] These mutations in serum surfactant protein C may damage type II alveolar epithelial cells. [23] Additionally, a common variant in the putative promoter of the gene encoding mucin 5B (MUC5B) has been associated with the development of both familial interstitial pneumonia and sporadic pulmonary fibrosis. [21]
Finally, mutant telomerase is associated with familial idiopathic pulmonary fibrosis. [20] Pulmonary fibrosis in patients with short telomeres is provoked by a loss of alveolar epithelial cells. Telomere shortening also occurs with aging and can also be acquired. This telomere shortening could promote the loss of alveolar epithelial cells, resulting in aberrant epithelial cell repair, and therefore should be considered as another potential contributor to the pathogenesis of idiopathic pulmonary fibrosis. [20]
Respiratory viruses have been considered a particularly likely cause of AE-IPF based on the similarities in clinical and radiologic presentation between and AE-IPF and viral pneumonitis and the poor sensitivity of standard methods of viral detection. A study by Wootton et al used genomics-based discovery methods to define the role of viral infections in AE-IPF. Initial multiplex polymerase chain reaction (PCR) revealed common respiratory viral infection in only 4 of 43 patients with AE-IPF. Pan-viral microarrays revealed torque teno virus (TTV) in 12 patients with AE-IPF. The pathogenic significance of TTV in AE-IPF is unclear. Overall, viral infection was not detected in most cases of AE-IPF. [24]
Epidemiology
United States
No large-scale studies of the incidence or prevalence of idiopathic pulmonary fibrosis (IPF) are available on which to base formal estimates.
In a retrospective administrative patient claim data study by Raghu et al in 2016, the annual cumulative prevalence of idiopathic pulmonary fibrosis in adults aged 18-64 years in the United States has increased from 13.4 cases per 100, 000 persons in 2005 to 18.2 cases per 100, 000 persons in 2010. [25]
A population-based cohort study was completed in Olmsted County, Minnesota, between 1997 and 2005, with the intention of updating and describing the incidence and prevalence of idiopathic pulmonary fibrosis. Narrow-criteria idiopathic pulmonary fibrosis was defined by usual interstitial pneumonia on a surgical lung biopsy specimen or a definite usual interstitial pneumonia pattern on an HRCT image. Broad-criteria idiopathic pulmonary fibrosis was defined by usual interstitial pneumonia on a surgical lung biopsy specimen or a definite or possible usual interstitial pneumonia pattern on an HRCT image. [26] These criteria were obtained from the 2002 American Thoracic Society/European Thoracic Society consensus statement. [10]
The age-adjusted and sex-adjusted incidence rate of idiopathic pulmonary fibrosis among residents aged 50 years or older ranges from 8.8 cases per 100,000 person-years (narrow-case criteria) to 17.4 cases per 100,000 person-years (broad-case criteria). [26]
The age-adjusted and sex-adjusted prevalence among residents aged 50 years or older ranges from 27.9 cases per 100,000 persons (narrow-case criteria) to 63 cases per 100,000 persons (broad-case criteria). [26]
Whether the incidence and prevalence of idiopathic pulmonary fibrosis are influenced by geographic, ethnic, cultural, or racial factors is unclear. [1]
International
Worldwide, the incidence of idiopathic pulmonary fibrosis is estimated to be 10.7 cases per 100,000 person-years for males and 7.4 cases per 100,000 person years for females. The prevalence of idiopathic pulmonary fibrosis is estimated to be 20 cases per 100,000 persons for males and 13 cases per 100,000 persons for females. [12]
Race
Epidemiologic data from large, geographically diverse populations are limited, and, therefore this data cannot be used to accurately determine the existence of a racial predilection for idiopathic pulmonary fibrosis.
Sex
Using data obtained from a large US healthcare claims database, the incidence and prevalence of idiopathic pulmonary fibrosis is higher in men aged 55 years or older, compared with women of the same age. [27]
Age
Idiopathic pulmonary fibrosis mainly affects persons aged 50 years or older. Approximately two thirds of persons diagnosed with idiopathic pulmonary fibrosis are aged 60 years or older at the time of diagnosis. Using data obtained from a large US healthcare claims database, the incidence of idiopathic pulmonary fibrosis was estimated to range from 0.4-1.2 cases per 100,000 person-years for persons aged 18-34 years. However, the estimated incidence of idiopathic pulmonary fibrosis in persons aged 75 years or older was significantly higher and ranged from 27.1-76.4 cases per 100,000 person-years. [27]
Prognosis
Idiopathic pulmonary fibrosis (IPF) portends a poor prognosis. With regard to idiopathic pulmonary fibrosis life expectancy, the estimated mean survival is 2-5 years from the time of diagnosis. [2] Estimated mortality rates are 64.3 deaths per million in men and 58.4 deaths per million in women. [28]
Death rates in patients with idiopathic pulmonary fibrosis increase with increasing age, are consistently higher in men than women, and experience seasonal variation, with the highest death rates occurring in the winter, even when infectious causes are excluded. [11]
Estimates are that 60% of patients with idiopathic pulmonary fibrosis die from their idiopathic pulmonary fibrosis, as opposed to dying with their idiopathic pulmonary fibrosis. Of those patients who die with idiopathic pulmonary fibrosis, most commonly it is after an acute exacerbation of idiopathic pulmonary fibrosis. When an acute exacerbation of idiopathic pulmonary fibrosis is not the cause of death, an increased cardiovascular risk and an increased venous thromboembolic disease risk contribute to the cause of death. The most common causes of death in patients with idiopathic pulmonary fibrosis include acute exacerbations of idiopathic pulmonary fibrosis, acute coronary syndromes, congestive heart failure, lung cancer, infectious causes, and venous thromboembolic disease. [2]
A worse prognosis can be expected based on various clinical parameters, physiologic factors, radiographic findings, histopathologic findings, laboratory findings, and bronchoalveolar lavage findings. du Bois et al evaluated a scoring system to predict individual risk of mortality. They used a Cox proportional hazards model and data from two clinical trials (n = 1,099) to identify independent predictors of 1-year mortality among patients with idiopathic pulmonary fibrosis. The findings demonstrated that 4 readily ascertainable predictors (age, history of respiratory hospitalization within the previous 24 weeks, percent predicted FVC, and 24-week change in FVC) could be used in a scoring system to estimate 1-year mortality. However, this scoring system needs to be validated in other populations of patients with idiopathic pulmonary fibrosis. [29]
Ley et al used competing risks regression modeling to retrospectively screen potential predictors of mortality in a derivation cohort of patients with idiopathic pulmonary fibrosis (n = 228). They identified a model consisting of 4 predictors (sex, age, % predicted FVC, and % predicted DLCO). Based on these 4 predictors, they developed a simple point-score model and staging system that was retrospectively validated in a separate cohort of patients with idiopathic pulmonary fibrosis (n = 330). [30]
Table 1. Scoring for mortality risk in IPF. (Open Table in a new window)
|
Predictor |
Points |
Sex |
Female |
0 |
Male |
1 |
|
Age (years) |
≥60 |
0 |
61-65 |
1 |
|
>65 |
2 |
|
FVC (% predicted) |
>75 |
0 |
50-75 |
1 |
|
< 50 |
2 |
|
DLCO (% predicted) |
>55 |
0 |
36-55 |
1 |
|
≤35 |
2 |
|
Cannot perform |
3 |
Table 2. Staging and mortality risk for IPF. (Open Table in a new window)
Stage |
I |
II |
III |
Points |
0-3 |
4-5 |
6-8 |
Mortality |
|
|
|
1-year |
5.6 |
16.2 |
39.2 |
2-year |
10.9 |
29.9 |
62.1 |
3-year |
16.3 |
42.1 |
76.8 |
The authors believe that the index and staging system provide clinicians with a framework for discussing prognosis, policy-makers with a tool for investigating stage-specific management options, and researchers with the ability to identify at-risk study populations that maximize the efficiency and power of clinical trials. [30]
Patients with idiopathic pulmonary fibrosis who have concomitant pulmonary hypertension have more dyspnea, greater impairment of their exercise capacity, and increased 1-year mortality compared with their counterparts without pulmonary hypertension. [2] Additionally, a multicenter prospective cohort study of 126 lung transplant procedures performed for idiopathic pulmonary fibrosis revealed elevated pulmonary artery pressure as a risk factor for primary graft dysfunction (PGD) following lung transplantation. [31] The mean pulmonary artery pressure (mPAP) for patients with PGD following lung transplantation was 38.5 ± 16.3 mm Hg compared with a mPAP of 29.6 ± 11.5 mm Hg in patients without PGD following lung transplantation.
Patients with idiopathic pulmonary fibrosis pattern on HRCT imaging have a worse prognosis compared with patients with biopsy-proven usual interstitial pneumonia and atypical changes of idiopathic pulmonary fibrosis on HRCT imaging. [11, 32]
Patients who have a greater than 10% decline in forced vital capacity (FVC) (percent predicted) over 6 months have a 2.4-fold increased risk of death. Additionally, in patients who do not desaturate to less than 88% during a 6-minute walk test (6MWT), the only strong predictor of mortality is a progressive decline in FVC (>10% after 6 mo). [33]
A baseline diffusion capacity of carbon monoxide (DLCO) below 35% is correlated with increased mortality. Additionally, a decline in DLCO greater than 15% over 1 year is also associated with increased mortality. [33]
Desaturation below the threshold of 88% during the 6MWT has been associated with an increased mortality. [33] Additionally, in patients with idiopathic pulmonary fibrosis who desaturate to less than 88% during a 6MWT, a progressive decline in DLCO (>15% after 6 mo) is a strong predictor of mortality. [7]
BAL fluid neutrophilia has been demonstrated to predict early mortality. One study demonstrated a linear relationship between increasing neutrophil percentage and the risk of mortality. Each doubling in baseline BAL fluid neutrophil percentage was associated with a 30% increased risk of death or transplantation in the first year after presentation. [34]
Serum surfactant protein A (SP-A) is a member of the collectin family. SP-A is secreted by type II pneumocytes, and the level of SP-A appears to be increased early after breakdown in the alveolar epithelium. SP-A has been shown to be present in abnormal amounts in the BAL fluid of patients with idiopathic pulmonary fibrosis. [35] In a cohort study, after controlling for known clinical predictors of mortality, each increase of 49 ng/mL in baseline serum SP-A level was associated with a 3.3-fold increased risk of mortality in the first year after presentation. [35] Therefore, serum SP-A is independently and strongly associated with death or lung transplantation 1 year after presentation. [35]
Patient Education
Patients should be presented information regarding the full range of options available for treating idiopathic pulmonary fibrosis (IPF). The pros, cons, risks, benefits, and alternatives should be discussed in a balanced and comprehensive fashion. For patient education resources, see the Lung Disease and Respiratory Health Center.
-
Chest radiograph of a patient with idiopathic pulmonary fibrosis showing bilateral lower lobe reticular opacities (red circles).
-
Classic subpleural honeycombing (red circle) in a patient with a diagnosis of idiopathic pulmonary fibrosis.
-
A patient with IPF and a confirmed histologic diagnosis of usual interstitial pneumonia. Note the reticular opacities (red circle) distributed in both lung bases and the minimal ground-glass opacities (blue circle).
-
A patient with nonspecific interstitial pneumonia. Note the predominance of ground-glass opacities (blue circles) and a few reticular lines (red arrow).
-
Patchwork distribution of abnormalities in a classic example of usual interstitial pneumonia (low-magnification photomicrograph; hematoxylin and eosin stain; original magnification, X4). Courtesy of Chad Stone, MD.
-
High-resolution CT coronal view showing lower lobe predominant honeycombing (blue arrow) with irregular septal thickening and traction bronchiectasis compatible with typical usual interstitial pneumonia pattern.
Tables
|
Predictor |
Points |
Sex |
Female |
0 |
Male |
1 |
|
Age (years) |
≥60 |
0 |
61-65 |
1 |
|
>65 |
2 |
|
FVC (% predicted) |
>75 |
0 |
50-75 |
1 |
|
< 50 |
2 |
|
DLCO (% predicted) |
>55 |
0 |
36-55 |
1 |
|
≤35 |
2 |
|
Cannot perform |
3 |
Stage |
I |
II |
III |
Points |
0-3 |
4-5 |
6-8 |
Mortality |
|
|
|
1-year |
5.6 |
16.2 |
39.2 |
2-year |
10.9 |
29.9 |
62.1 |
3-year |
16.3 |
42.1 |
76.8 |
