Overview
Torsade de pointes is an uncommon and distinctive form of polymorphic ventricular tachycardia (VT) characterized by a gradual change in the amplitude and twisting of the QRS complexes around the isoelectric line (see the image below). Torsade de pointes, often referred to as torsade, is associated with a prolonged QT interval, which may be congenital or acquired. [1] Torsade usually terminates spontaneously but frequently recurs and may degenerate into ventricular fibrillation, and therefore can be fatal if not diagnosed and managed.
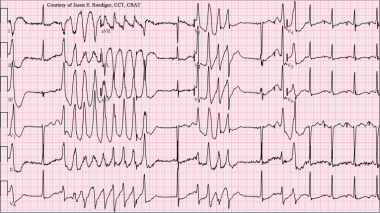 Torsade de Pointes. Torsades de pointes with prolonged QT interval (12-lead ECG of torsades de pointes (TdP) in a 56-year-old white female with low blood potassium (2.4 mmol/L) and low blood magnesium (1.6 mg/dL). Courtesy of Wikimedia Commons [Author Jason E Roediger, CCT, CRAT, https://commons.wikimedia.org/wiki/File:Torsades_de_Pointes_TdP.png].
Torsade de Pointes. Torsades de pointes with prolonged QT interval (12-lead ECG of torsades de pointes (TdP) in a 56-year-old white female with low blood potassium (2.4 mmol/L) and low blood magnesium (1.6 mg/dL). Courtesy of Wikimedia Commons [Author Jason E Roediger, CCT, CRAT, https://commons.wikimedia.org/wiki/File:Torsades_de_Pointes_TdP.png].
In torsade, the morphology of the QRS complexes varies from beat to beat. The ventricular rate can range from 150 beats per minute (bpm) to 250 bpm.
The original report described regular variation of the morphology of the QRS vector from positive to net negative and back again. This was symbolically termed torsade de pointes, or "twisting of the point" about the isoelectric axis, because it reminded the authors of the torsade de pointes movement in ballet. Most cases exhibit polymorphism, but the axis changes may not have regularity.
The definition also requires that the QT interval be increased markedly (often to ≥600 msec). Cases of polymorphous ventricular tachycardia in which the QT interval is not prolonged are treated as generic ventricular tachycardia.
Pathophysiology
The association between torsade and a prolonged QT interval has long been known, but the mechanisms involved at the cellular and ionic levels have been made clearer in approximately the last decade. The abnormality underlying both acquired and congenital long QT syndromes is in the ionic current flow during repolarization, which affects the QT interval.
A variety of changes in ionic current can result in the common effect of decreased repolarizing current, reflected in a long QT, and these changes can secondarily lead to subsequent depolarizing currents and sometimes action potentials, termed afterdepolarizations. This leads to a further delay in repolarization and causes early afterdepolarization (EAD), the triggering event for torsade. [2]
Repolarization has three phases. During the initial upstroke of action potential in a normal cardiac cell, a rapid net influx of positive ions (Na+ and Ca++) occurs, which results in the depolarization of the cell membrane. This is followed by a rapid, transient outward potassium current (Ito), while the influx rate of positive ions (Na+, Ca++) declines. This represents the initial part of the repolarization, or phase 1.
Phase 2 is characterized by the plateau. The positive currents flowing inward and outward become almost equal during this stage.
Phase 3 of repolarization is mediated by activation of the delayed rectifier potassium current (IK) moving outward while the inward positive current decays. If a slow inactivation of the Ca++ and Na+ currents occurs, this inward "window" current can cause single or repetitive depolarization during phases 2 and 3 (ie, EADs). These EADs appear as pathologic U waves on a surface ECG, and, when they reach a threshold, they may trigger ventricular tachyarrhythmias.
These changes in repolarization do not occur in all myocardial cells. The deep endocardial region and midmyocardial layer (composed of M cells) of the ventricle are more prone to prolongation of repolarization and EADs because they have a less-rapid delayed rectifier potassium current (IKr), while other regions might have short or normal cycles. This heterogeneity of repolarization in the myocardial cells promotes the spread of triggered activity, which is initiated by EADs by a reentrant mechanism and currently is thought to be responsible for the maintenance of torsade. [3]
Six genetic variants underlying torsade are currently recognized. Genotypes LQT1 and LQT2 have slow potassium channels, while LQT3 shows defects in the sodium channels. Treatment modalities soon may be based on the genotype of the individual.
Etiology of Torsade
Prolongation of the QT interval may be congenital, as seen in the Jervell and Lange-Nielsen syndrome (ie, congenitally long QT associated with congenital deafness) and the Romano Ward syndrome (ie, isolated prolongation of QT interval). Both of these syndromes are associated with sudden death due to either primary ventricular fibrillation or torsade that degenerates into ventricular fibrillation.
Brugada syndrome is characterized by a coved ST segment in the right precordial leads. The syndrome may cause sudden death due to polymorphic ventricular tachycardia resembling torsade.
Takotsubo cardiomyopathy (stress-induced cardiomyopathy) causes a predisposition to torsade. [4, 5]
The acquired conditions that predispose one to torsade either decrease the outward potassium current or interfere with the inward sodium and calcium currents, or fluxes.
The electrolyte disturbances that have been reported to precipitate torsade include hypokalemia [6] and hypomagnesemia. These abnormalities cause a delay in phase III (ie, reprolongation) and form the substrate for emergence of the dysrhythmia. Close observation is required in predisposed patients, such as those with cirrhosis or hypothyroidism.
Antiarrhythmic drugs reported to be etiologic include class IA agents (eg, quinidine, procainamide, disopyramide), class IC agents (eg, flecainide [encainide was withdrawn in 1991]), and class III agents (eg, sotalol, amiodarone).
Other drugs that prolong the QT interval and have been implicated in cases of torsade include phenothiazines, tricyclic antidepressants, lithium carbonate, ziprasidone, [7] cisapride, highly active antiretroviral drugs, high-dose methadone, anthracycline chemotherapeutic agents (eg, doxorubicin, daunomycin), [8] some fluoroquinolones, and any other medication using the CYP3A metabolic pathway. Ranolazine, [9] an antiangina agent, also prolongs the QTc, but torsade is a rare complication of this therapy. Often, multiple agents act synergistically. Because QT prolongation is highly variable among and even in a single individual, the specific "causative" agent is often speculative. Accordingly, lists of agents suspected of increasing the QT interval may include some drugs that do not have this particular effect.
A single-center, retrospective (2008-2019), observational study of the effects of 272 medications on 310,335 electrocardiograms (ECGs) from 159,397 individuals found that drugs associated with the greatest QTc prolongation were dofetilide, mexiletine, amiodarone, rifaximin, and sotalol. [10] However, the investigators also noted that several top drugs not previously known to prolong QT included rifaximin, lactulose, cinacalcet, and lenlidomide.
A 2023 case report described emergency department administration of a small dose (4 mg) of intravenous ondansetron resulting in QT prolongation, torsade, and cardiac arrest in a middle-aged woman with alcohol disuse disorder who was not on any medications or supplements. [11] Another 2023 case report described a rare presentation of levetiracetam-induced torsade and cardiac arrest in a middle-age man with a history of a seizure disorder but noncompliance with valproic acid. [12] The patient received a total of 1000 mg intravenous levetiracetam as well as 4 mg of intramuscular lorazepam for two sets of episodes of tonic-clonic seizures in the emergency department.
Complications from SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) infection itself as well as from its associated treatment, including with remdesivir, can also precipitate prolonged QTc and torsade de pointes. [13]
Drug interactions with the antihistamines astemizole (recalled from US market) and terfenadine (recalled from US market) can precipitate torsade; these drugs should never be used with class IA, IC, or III agents. Astemizole and terfenadine, in high dosages or when used in combination with the azole antifungal drugs or the macrolide antibiotics, have been reported to precipitate torsade and sudden death. Grapefruit juice has been shown to slow the hepatic metabolism of these antihistamines as well as other drugs and to prolong the QT interval in patients taking astemizole or terfenadine. Clinical implications of this interaction are unclear.
Risk factors
Risk factors for torsade include:
-
Congenital long QT syndrome
-
Female gender
-
Acquired long QT syndrome (causes of which include medications and electrolyte disorders such as hypokalemia and hypomagnesemia)
-
Bradycardia
-
Baseline electrocardiographic abnormalities
-
Renal or liver failure
Congenital long QT syndromes (adrenergic-dependent)
Congenital syndromes associated with torsade include:
-
Jervell and Lange-Nielsen syndrome
-
Romano-Ward syndrome
Acquired long QT syndromes
Drugs in a number of drug classes have been associated with torsade.
Antiarrhythmic drugs associated with torsade include:
-
Class IA: Quinidine, disopyramide, procainamide
-
Class III: Sotalol, amiodarone (rare), ibutilide, dofetilide, almokalant
Other drug classes associated with torsade include:
-
Antibiotics: Erythromycin, clarithromycin, azithromycin, levofloxacin, moxifloxacin, gatifloxacin, trimethoprim-sulfamethoxazole, clindamycin, pentamidine, chloroquine
-
Antifungals: Ketoconazole, itraconazole
-
Antivirals: Amantadine
-
Antipsychotics: Haloperidol, phenothiazines, thioridazine, trifluoperazine, sertindole, zimeldine, ziprasidone [14]
-
Tricyclic and tetracyclic antidepressants
-
Antihistamines (histamine1-receptor antagonists): Diphenhydramine, hydroxyzine [15] (Terfenadine and astemizole were recalled from the US market.)
-
Cholinergic antagonists: Cisapride, organophosphates (pesticides)
-
Diuretics: Indapamide, hydrochlorothiazide, furosemide
-
Antihypertensives: Bepridil, lidoflazine, prenylamine, ketanserin
-
Lithium
-
Anticonvulsants: Phenytoin, carbamazepine (possible)
-
Oral hypoglycemic
-
Citrate (massive blood transfusions)
-
Cocaine
-
Vasopressin (possible)
-
Fluoxetine (possible)
Some drugs (eg, amiodarone) routinely prolong QT but are less commonly associated with clinical consequences of long QT.
Conditions associated with torsade include:
-
Electrolyte abnormalities: Hypokalemia, hypomagnesemia, hypocalcemia
-
Endocrine disorders: Hypothyroidism, hyperparathyroidism, pheochromocytoma, hyperaldosteronism, hypoglycemia
-
Cardiac conditions: Myocardial ischemia, myocardial infarction, myocarditis, bradyarrhythmia, complete atrioventricular (AV) block, [20] takotsubo cardiomyopathy
-
Intracranial disorders: Subarachnoid hemorrhage, thalamic hematoma, cerebrovascular accident, encephalitis, head injury
-
Nutritional disorders: Anorexia nervosa, starvation, liquid protein diets, gastroplasty and ileojejunal bypass, celiac disease
Relatively recent studies on out-of-hospital cardiac arrest demonstrate that certain prescription medications (particularly antidepressants and antipsychotics) are associated with a nonshockable rhythm. [21] A follow-up study concluded that use of drugs known to increase torsade risk (most commonly citalopram and roxithromycin) was associated with a reduced likelihood of presenting with a shockable rhythm and thus a less likely to return to spontaneous circulation. [22]
Epidemiology of Torsade
The prevalence of torsade de pointes is unknown. Torsade is a life-threatening arrhythmia and may present as sudden cardiac death in patients with structurally normal hearts. [23] In the United States, 300,000 sudden cardiac deaths occur per year. Torsade probably accounts for fewer than 5%.
For both sexes, the corrected QT interval is longer in White persons than in Black persons, possibly explaining the lower susceptibility to acquired torsade in Black persons. Brugada syndrome is more frequent in Southeast Asians. [24]
Torsade is 2-3 times more common in women than in men. Women have longer QT intervals, [25] as well as have more QT prolongation secondary to drug therapy. Congenital long QT syndrome is autosomal in genetic transmission but shows a greater frequency of expression and a greater lengthening of the QT interval in women than in men.
Torsade occurs in patients of a wide age range, from newborns to the very elderly. The highest frequency is in patients aged 35-50 years. Torsade that occurs at an early age usually is due to congenital long QT syndrome. In older persons, it usually is due to acquired long QT syndrome. In a systematic review, investigators found that slightly over half (50.8%) of elderly patients (age ≥80 years) with drug-induced torsade experienced it as the result of "reckless administration" of a QT-interval prolonging agent. [26] The most common occurrences of reckless administration of a QT-interval prolonging were in conjunction with another such agent (51.6%) or despite a known QT-interval prolongation (25.8%). [26]
Presentation
Patient history
Patients with torsade usually present with recurrent episodes of palpitations, dizziness, and syncope that correspond to arrhythmia episodes; however, sudden cardiac death can occur with the first episode. Nausea, cold sweats, shortness of breath, and chest pain also may occur but are nonspecific and can be produced by any form of tachyarrhythmia.
In a young patient with torsade, a diagnosis of congenital long QT syndrome should be considered, especially if a family history of sudden cardiac death or sudden infant death syndrome is present. In these patients, episodes of torsade are triggered by adrenergic stimulation such as stress, fear, or physical exertion, [27] but other predisposing factors also should be considered. See Long QT Syndrome.
A family history of congenital deafness may also be suggestive, although a prolonged QT is found in only 0.25-0.3% of deaf-mute children. Patients with Jervell and Lange-Nielsen syndrome commonly have congenital sensorineural deafness representing an autosomal dominant pattern of inheritance for cardiac abnormalities, whereas deafness usually is autosomal recessive.
Another form of familial or congenital long QT syndrome is Romano-Ward syndrome, in which hearing is normal and an autosomal dominant pattern of inheritance is observed.
Patients with acquired long QT syndrome usually develop torsade during periods of bradycardia. The most common causes of acquired long QT syndrome are medications and electrolyte disorders (eg, hypokalemia, hypomagnesemia). Drug-associated torsade de pointes is relatively rare, but is becoming increasingly common; its incidence is as high as 2-3% with certain drugs. Hence, asking the patient about all current medications is important.
Physical examination
The physical findings in torsade depend on the rate and duration of tachycardia and the degree of cerebral hypoperfusion.
Findings include rapid pulse, low or normal blood pressure, or transient or prolonged loss of consciousness. This could be preceded by bradycardia or premature ventricular contractions (leading palpitations). An increase in premaure ventricular contractions in individuals with prolonged QTc may be useful in predicting imminent torsade. [28]
Pallor and diaphoresis may be noted, especially with a sustained episode.
Other physical signs depend on the etiology of torsade.
Differential Diagnosis for Torsade
The differential diagnosis of torsade de pointes includes:
Other considerations are the differentiation of acquired long QT syndrome from congenital long QT syndrome. In addition, torsade should be differentiated from polymorphic ventricular tachycardia or, rarely, monomorphic ventricular tachycardia.
Supraventricular tachycardia with aberrant conduction may be confused with torsade, especially when the degree of aberration is variable. One clue to the former is that atrial fibrillation may be intermixed with narrower and typical QRS complexes.
Electrocardiography
Torsade is an electrocardiographic (ECG) diagnosis, and obtaining an ECG is essential. [29] Typical examples are depicted below.
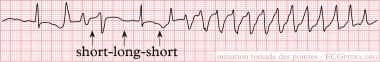 Torsade de Pointes. An example of torsade de pointes. A normal sinus beat is followed by a ventricular extrasystole (shortly after the sinus beat), the compensatory pause results in a longer interval to the next beat. This longer beat therefore has a longer QT interval. The next beat follows shortly thereafter, within the QT interval. Not all ventricular cells have been repolarized by that time and a ventricular arrhythmia results. This short-long-short sequence is typical for torsade de pointes. Courtesy of Wikimedia Commons [Author CardioNetworks, https://commons.wikimedia.org/wiki/File:Rhythm_torsade_(CardioNetworks_ECGpedia).png].
Torsade de Pointes. An example of torsade de pointes. A normal sinus beat is followed by a ventricular extrasystole (shortly after the sinus beat), the compensatory pause results in a longer interval to the next beat. This longer beat therefore has a longer QT interval. The next beat follows shortly thereafter, within the QT interval. Not all ventricular cells have been repolarized by that time and a ventricular arrhythmia results. This short-long-short sequence is typical for torsade de pointes. Courtesy of Wikimedia Commons [Author CardioNetworks, https://commons.wikimedia.org/wiki/File:Rhythm_torsade_(CardioNetworks_ECGpedia).png].
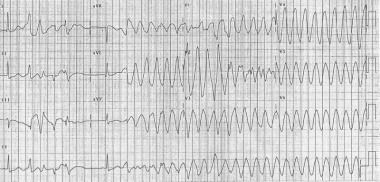 Torsade de Pointes. 12 lead Torsade de Pointes. Courtesy of Wikimedia Commons [Author CardioNetworks, https://commons.wikimedia.org/wiki/File:12leadTorsade_(CardioNetworks_ECGpedia).jpg].
Torsade de Pointes. 12 lead Torsade de Pointes. Courtesy of Wikimedia Commons [Author CardioNetworks, https://commons.wikimedia.org/wiki/File:12leadTorsade_(CardioNetworks_ECGpedia).jpg].
Frequent ECG monitoring is indicated for patients who are at risk due to chronic conditions or drug therapy. When the patient is in sinus rhythm, examine the QT interval. Usually, a prolonged QT interval and pathological U waves are present, reflecting abnormal ventricular repolarization. The most consistent indicator of QT prolongation is a QT of 0.60 s or longer or a QTc (corrected for heart rate) of 0.45 s or longer.
Other electrocardiographic features helpful in diagnosing torsade include its typical mode of onset and its morphology, as follows:
-
Patients have paroxysms of 5-20 beats at a rate faster than 200 bpm; sustained episodes occasionally can be seen
-
Progressive change in polarity of QRS about the isoelectric line occurs
-
Complete 180° twist of QRS complexes in 10-12 beats is present
-
A short-long-short sequence between the R-R intervals occurs before the trigger response.
-
Patients may revert spontaneously or convert to a nonpolymorphic ventricular tachycardia or ventricular fibrillation
-
Occasionally, T-wave alternans may be seen before torsade
Torsade occurring in the setting of acquired long QT syndrome is preceded by pauses in almost all cases. In congenital long QT syndrome (adrenergic-dependent), pause dependence is found in most of the adult cases, whereas onset of torsade is not pause-dependent in children.
Failure to identify this rhythm may occur for various reasons. During very short runs of torsade, the typical oscillatory QRS complexes around the isoelectric line may not be apparent. Early events usually are short lived. In the case of a single-lead recording, the typical morphology of torsade may not be obvious.
The diagnosis of torsade should be considered in any patient with pause-dependent ventricular tachycardia, and ventricular bigeminy in a patient with long QT interval may be a sign of an impending torsade.
Findings from electrophysiological studies usually are negative in torsade.
Other Tests
Other tests, including lab and imaging studies, should be ordered based on the etiological factors being considered (see Etiology of Torsade).
Laboratory studies
Electrolytes: Check for hypoglycemia, hypokalemia, hypomagnesemia, and hypocalcemia.
Cardiac enzymes: Rule out myocardial ischemia, especially in patients without QT prolongation.
Imaging studies
Chest radiographs and echocardiography should be performed to rule out structural heart disease, if any clinical suggestion is present.
Treatment of Torsade
Consultations
Immediate cardiology evaluation and follow-up are required. Other possible consultations include:
-
Electrophysiologist
-
Cardiologist
-
Geneticist (in cases of familial or congenital long QT syndrome)
Acute management
Treatment of torsade de pointes can be divided into short-term and long-term management. The goal of short-term management is to assess for, achieve, and maintain hemodynamic stability. Short-term management of torsade is the same in both acquired and congenital long QT syndrome, except that beta1-adrenergic stimulation may be tried in the acquired form but is contraindicated in the congenital form.
In an otherwise stable patient, direct current (DC) cardioversion is kept as a last resort, because torsade is paroxysmal in nature and is characterized by its frequent recurrences following cardioversion. Although torsade frequently is self-terminating, it may degenerate into ventricular fibrillation, which requires DC defibrillation.
Any offending agent should be withdrawn. Predisposing conditions such as hypokalemia, hypomagnesemia, and bradycardia should be identified and corrected.
Pharmacologic therapy
Magnesium is the drug of choice for suppressing early afterdepolarizations (EADs) and terminating the arrhythmia. Magnesium achieves this by decreasing the influx of calcium, thus lowering the amplitude of EADs. [30]
Magnesium can be given at 1-2 g IV initially in 30-60 seconds, which then can be repeated in 5-15 minutes. Alternatively, a continuous infusion can be started at a rate of 3-10 mg/min. Magnesium is effective even in patients with normal magnesium levels. Because of the danger of hypermagnesemia (depression of neuromuscular function), the patient requires close monitoring.
Some authorities recommend supplemental potassium to increase the potassium concentration to high normal, which increases the efflux of potassium from myocardial cells, thus causing rapid repolarization.
Lidocaine usually has no effect in torsade. Occasionally, it can have an initial beneficial effect, but torsade recurs in all cases.
Mexiletine also may be helpful in suppressing torsade. In one study, it was used in patients with HIV who had acquired long QT interval and torsade. [31] It effectively suppressed the torsade on a long-term basis. Nakashima et al reported successfully preventing refractory torsade de pointes and ventricular fibrillation in a 28-year-old patient with congenital type 2 long QT syndrome. [32]
Patients with congenital long QT syndromes are thought to have an abnormality of sympathetic balance or tone and are treated with beta-blockers. If the patient experiences breakthrough torsade, a short-acting beta-blocker, such as esmolol, can be tried. [33]
Isoproterenol can be used in bradycardia-dependent torsade that usually is associated with acquired long QT syndrome (pause-dependent). It should be administered as a continuous IV infusion to keep the heart rate above 90 bpm.
Isoproterenol accelerates AV conduction and decreases the QT interval by increasing the heart rate and reducing temporal dispersion of repolarization. Beta-adrenergic agonists such as isoproterenol are contraindicated in the congenital form of long QT syndrome (adrenergic-dependent). Because of precautions, contraindications, and adverse effects associated with its use, this drug is used as an interim agent until overdrive pacing can be started.
Temporary transvenous pacing
Based on the fact that the QT interval shortens with a faster heart rate, pacing can be effective in terminating torsade. It is effective in both forms of the long QT syndrome because it facilitates the repolarizing potassium currents and prevents long pauses, suppressing EADs and decreasing the QT interval.
Atrial pacing is the preferred mode because it preserves the atrial contribution to ventricular filling and also results in a narrower QRS complex and hence a shorter QT. In patients with AV block, ventricular pacing can be used to suppress torsade. This is dependent on intact atrial-to-ventricular conduction at the pacing rate found necessary.
Pacing should be instituted at a rate of 90-110 bpm until the QT interval is normalized. Overdrive pacing may be necessary at a rate of up to 140 bpm to control the rhythm.
The patient with torsade who is in extremis should be treated with electrical cardioversion or defibrillation. Anecdotal reports cite successful conversion with phenytoin (Dilantin) and lidocaine. A few cases of successful conversion using phenytoin and overdrive pacing have been reported.
If patient is unresponsive to conversion with phenytoin and overdrive pacing, attempt electrical cardioversion.
Long-term treatment
Beta-adrenergic antagonists at maximally tolerated doses are used as a first-line long-term therapy in congenital long QT syndrome. Propranolol is used most extensively, but other agents such as esmolol or nadolol also can be used. Beta-blockers should be avoided in those congenital cases in which bradycardia is a prominent feature. Beta-blockers are contraindicated in acquired long QT syndrome because bradycardia produced by these agents can precipitate torsade. One approach to assess the adequacy of beta-blockade is by exercise testing. One investigator recommends aiming for at least a 20% reduction in maximum heart rate compared to that of the baseline (pre-beta blocker therapy). Another approach is to check the blood levels of beta blockers (eg, propranolol) when possible. [34]
Patients without syncope, ventricular tachyarrhythmia, or a family history of sudden cardiac death can be observed without starting any treatment.
Permanent pacing benefits patients who remain symptomatic despite receiving the maximally tolerated dose of beta-blockers and can be used adjunctively with beta-blockers. It decreases the QT interval by enhancing the repolarizing potassium currents and suppressing EADs.
High left thoracic sympathectomy, another antiadrenergic therapy, is effective in patients who remain refractory to beta-blockade and pacing. Accidental ablation of ocular efferent sympathetic nerves may result in Horner syndrome.
Implantable cardioverter-defibrillators (ICDs) are useful in instances when torsade recurs despite treatment with beta-blockers, pacing, and possibly left thoracic sympathectomy. Beta-blockers should be used along with ICDs because shock can further precipitate torsade by adrenergic stimulation. In the United States, an ICD for refractory cases may often precede sympathectomy.
Long-term treatment in acquired long QT syndrome usually is not required because the QT interval returns to normal once the inciting factor or predisposing condition has been corrected. Pacemaker implantation is effective in cases that are associated with heart block or bradycardia. ICDs are indicated in cases that cannot be managed by avoidance of the offending agent.
The boundary between acquired and congenital may not always be clear. Additive factors are often present, and individuals may show increased susceptibility to QT effects.
Activity
Competitive sports are prohibited in patients with congenital long QT syndrome.
Complications
Complications of torsade may include:
-
Monomorphic ventricular tachycardia
-
Ventricular fibrillation
-
Sudden cardiac death
Prognosis
In congenital long QT syndrome, the mortality rate for untreated patients is 50% in 10 years, which can be reduced to 3-4% with therapeutic intervention.
A systematic review of fetal long QT syndrome from 83 studies comprising 265 fetuses with postnatal confirmation of long QT syndrome found that a longer fetal QTc was more predictive of death than any other antenatal factor, and the mortality risk was significantly raised when the fetal QTc was longer than 600 ms. [35] Other factors that were highly predictive of mortality included the combination of ventricular tachycardia/torsade de pointes or functional 2:1 heart block and lack of a family history of long QT syndrome. However, fetal heart rate and heart z-score did not predict death. [35]
In acquired long QT syndrome, the prognosis is excellent once the inciting factor has been identified and reliably withheld.
Patient education
Instruct patients to use medications only with the approval of a physician.
Instruct patients to avoid competitive sports (in cases of congenital long QT syndrome).
Close follow-up is needed because of a risk of sudden cardiac death. Offer emotional support; suggest attending a cardiac support group.
Patients should be taught how to monitor their pulse and recognize adverse drug effects. Families should undergo training for basic life support.
Prevention of Torsade
Increased awareness of congenital QT syndrome and systematic strategies to assess, monitor, and manage patients with drug-induced QTc prolongation may help prevent this rare yet serious syndrome. A 2019 literature-based algorithm with a stepped-based approach for the assessment, monitoring, and management of drug-induced QTc prolongation in the psychiatric population is a good example of a tool that pharmacists can use to help mental health clinicians. [21]
Due to rapidly changing and expanding data on drugs known to increase QTc, a clinical decision support tool at the prescribing point of care via electronic health records offers an important strategy for prevention of torsade de pointes. [36, 37, 38, 39]
The Tisdale QT risk score assigns weighted values to various risk factors (including age, sex, hypokalemia, admission QTc interval, use of a loop diuretic, number of QT-prolonging drugs, sepsis, heart failure, and admission for acute myocardial infarction) and then estimates the risk of QT prolongation. Alerts to clinicians with incorporation of the the Tisdale QT risk reduced the prescribing of medications known to prolong QTc and lowered the occurrence of excessive QT prolongation in cardiac care units. [37] Upstream decision support tools in outpatient settings to highlight the risk of QT prolongation may help further reduce the risk of acquired long QT syndrome and thus the risk of torsade.
2023 CCS Clinical Practice Update on Management of Prolonged QT Interval
In October 2023, the Canadian Cardiovascular Society (CCS) released their clinical practice update on managing patients with a prolonged QT interval. [1] A summary of their practical tips are outlined below.
QT interval practice tips
It is recommended that the QT interval be measured manually owing to the potential for inaccuracy with automated measurements of the absolute QT interval (QTa) and the QT interval corrected for heart rate (QTc) in the setting of an irregular rhythm, widened QRS, or an abnormal ST-T wave configuration. This is due to the computer having difficulty defining the end of the T wave.
Manual QT measurement, ideally in leads V5 or II, involves using the maximal slope or tangen method for defining the end of the T wave.
Immediate post-cardiac arrest QT interval measurements may be unreliable due to cooling and myocardial and central nervous system injury. To avoid an overdiagnosis of long QT syndrome (LQTS), obtain serial electrocardiograms (ECGs).
Look for QT prolongation in the setting of unexplained syncope, seizures, or resuscitated cardiac arrest, or when there is a family history of unexplained sudden death.
When no limitations are present to accurately measure QTc, a QTc of 480 ms or longer is typically considered clearly abnormal; a QTc of 500 ms or longer is associated with a greater risk of ventricular arrhythmia.
In interpreting a mildly prolonged QTc in an otherwise normal ECG, clinical correlation is important. Are there associated clinical factors present (eg, high-risk symptoms, family history of cLQTS, use of QT-prolonging agents, electrolyte abnormalities)?
In the setting of an apparently normal QTc interval but a history that suggests congenital LQTS (cLQTS) (eg, family history), consider more studies (eg, exercise stress testing, 4-minute recovery ECG) to detect latent abnormal repolarization (LQTS with concealed QT prolongation). Abnomal T wave morphology is also associated with cLQTS.
cLQTS
Diagnosis and genetic testing
Refer individuals with suspected or confirmed cLQTS to an inherited arrhythmia or cardiogenic clinic.
In the setting of cLQTS, a normal or borderline abnormal QTC interval is common (concealed QT prolongation) and cannot be used to rule out cLQTS. An abnormal T wave morphology is often present in these patients.
The diagnostic tests of choice for revealing QT prolongation in cLQTS are exercise stress testing and particularly the 4-minute recovery ECG. Clearly indicate the test is to "rule out LQTS—look for abnormal QT prolongation especially on the standing and 4-minute post exercise ECG."
Many patients with cLQTS will never manifest symptoms. Nonetheless, they should avoid QT-prolonging agents.
Although there exist three autosomal dominant genes with strong evidence for cLQTS (and 14 uncommon genes that are less compelling), some individuals with clinical cLQTS do not have a recognized variant (genetically elusive LQTS).
In the setting of identification of a causative variant in a proband, offer first-degree relatives genotype-based cascade screening along with clinical phenotyping. In the setting of a genotype-negative proband, offer first-degree relatives screening via clinical evaluation (history, resting ECG, treadmill testing).
Management
Beta blockade (nadolol or propanolol is preferred) is the mainstay of medical therapy. It is rare that cardiac rhythm devices (eg, pacemakers, implantable cardioverter-defibrillators [ICDs]) are needed.
To improve tolerance of beta blockade, slowly uptitrate with the final dosage based on tolerance or blunted exercise heart rate.
Patients should avoid QT-prolonging agents. The majority of torsade de pointes episodes are from noncompliance with beta blockade or the use of QT-prolonging medications.
Manage torsade de pointes and arrhythmic storm with beta blockage, cessation of QT-prolonging agents, correction of electrolyte abnormalities, increased magnesium intake, lidocaine and, at times, transvenous pacing. Do not use amiodarone and procainamide (both have QT-interval prolongation and bradycardia-promoting properties).
Acquired LQTS (aLQTS)
The classic aLQTS is from the use of QT-prolonging agents (antidepressants/antipsychotics, antibiotics, antiarrhythmics) or electrolyte changes. Before administering medication, look for complex drug interactions or potential QT prolongation. Additionally, consider obtaining an ECG before starting a QT-prolonging drug in the presence of an electrolyte anomaly, if the patient is already on a QT-prolonging drug, or when starting a high-risk QT-prolonging agent.
Use caution when multiple risk factors for QT prolongation exist in a patient.
Note that important intraclass differences exist among QT-prolonging drugs in their risk for causing an arrhythmia. For example, this risk among macrolide antibiotics is greatest with erythromycin and least with azithromycin. Among selective serotonin reuptake inhibitors, the risk of arrhythmia is greatest with citalopram and escitalopram but neglible with paroxetine and sertraline. Stimulants used to treat attention deficit–hyperactivity disorder are generally considered to be safe.
The risk for torsade de pointes rises when the QTc is over 500 ms or when a medication increases the QTc by more than 60-70 ms, especially when there is a rapid increase.
Women, the elderly, and those with underlying structural heart disease have a higher risk of aLQTS.
Rule out underlying cLQTS if the QTc does not normalize after removing or correcting the inciting cause.
Pediatric population
Serial ECGs may be needed to make a diagnosis of LQTS in children because of challenges in measuring their QT interval (faster heart rates, sinus arrhythmia, age and hormonal factors). Note, however, that serial ECGs and overdiagnosis of cLQTS may result from reflex or vasovagal/vasodepressor syncope that is common in the prepubertal rapid growth phase.
Management of cLQTS in most pediatric patients can be achieved with beta blockers and avoidance of QT-prolonging drugs, and it is rare that cardiac rhythm devices are needed. Underdosing or missed doses of beta blockers or prescribed QT-prolonging agents are the predominant cause of breakthrough arrhythmic events.
Physical activity and sports participation can be encouraged with safety planning, such as access to an automated external defibrillator (AED) and supervision, particularly around swimming.
Prior to initiating genetic testing, obtain expert guidance to allow for appropriate informed consent and guidance on response to positive results.
-
Torsade de Pointes. Torsades de pointes with prolonged QT interval (12-lead ECG of torsades de pointes (TdP) in a 56-year-old white female with low blood potassium (2.4 mmol/L) and low blood magnesium (1.6 mg/dL). Courtesy of Wikimedia Commons [Author Jason E Roediger, CCT, CRAT, https://commons.wikimedia.org/wiki/File:Torsades_de_Pointes_TdP.png].
-
Torsade de Pointes. An example of torsade de pointes. A normal sinus beat is followed by a ventricular extrasystole (shortly after the sinus beat), the compensatory pause results in a longer interval to the next beat. This longer beat therefore has a longer QT interval. The next beat follows shortly thereafter, within the QT interval. Not all ventricular cells have been repolarized by that time and a ventricular arrhythmia results. This short-long-short sequence is typical for torsade de pointes. Courtesy of Wikimedia Commons [Author CardioNetworks, https://commons.wikimedia.org/wiki/File:Rhythm_torsade_(CardioNetworks_ECGpedia).png].
-
Torsade de Pointes. 12 lead Torsade de Pointes. Courtesy of Wikimedia Commons [Author CardioNetworks, https://commons.wikimedia.org/wiki/File:12leadTorsade_(CardioNetworks_ECGpedia).jpg].
