Practice Essentials
Dilated cardiomyopathy (DCM) is a progressive disease of heart muscle that is characterized by ventricular chamber enlargement and contractile dysfunction. The right ventricle may also be dilated and dysfunctional. DCM is the third most common cause of heart failure (HF) and the most frequent reason for heart transplantation.
Signs and Symptoms
Symptoms are a good indicator of the severity of DCM and may include the following:
-
Fatigue
-
Dyspnea on exertion, shortness of breath, cough
-
Orthopnea, paroxysmal nocturnal dyspnea
-
Increasing edema, weight, or abdominal girth
On physical examination, look for signs of HF and volume overload. Assess vital signs with specific attention to the following:
-
Tachypnea
-
Tachycardia
-
Hypertension or hypotension
Other pertinent findings include the following (the level of cardiac compensation or decompensation determines which signs are present):
-
Signs of hypoxia (eg, cyanosis, clubbing)
-
Jugular venous distension (JVD)
-
Pulmonary edema (crackles and/or wheezes)
-
S3 gallop
-
Enlarged liver
-
Ascites or peripheral edema
Look for the following on examination of the neck:
-
Jugular venous distention (as an estimate of central venous pressure)
-
Hepatojugular reflux
-
a wave on central venous pressure waveform
-
Large cv wave (observed with tricuspid regurgitation)
-
Goiter if dysthyroidism is suspected
Findings on examination of the heart may include the following:
-
Cardiomegaly (broad and displaced point of maximal impulse, right ventricular heave)
-
Murmurs (with appropriate maneuvers)
-
S2 at the base (paradoxical splitting, prominent P2), S3, and S4
-
Tachycardia
-
Irregularly irregular rhythm
-
Gallops
See Presentation for more detail.
Diagnosis
The workup in a patient with suspected cardiomyopathy may include the following:
-
Complete blood count
-
Comprehensive metabolic panel
-
Thyroid function tests
-
Cardiac biomarkers
-
B-type natriuretic peptide assay
-
Chest radiography
-
Echocardiography
-
Cardiac magnetic resonance imaging (MRI)
-
Electrocardiography (ECG)
In many cases of cardiomyopathy, endomyocardial biopsy is class II (uncertain efficacy and may be controversial) or class III (generally not indicated). Class II indications for endomyocardial biopsy include the following:
-
Recent onset of rapidly deteriorating cardiac function
-
Patients receiving chemotherapy with doxorubicin
-
Patients with systemic diseases with possible cardiac involvement (eg, hemochromatosis, sarcoidosis, amyloidosis, Löffler endocarditis, endomyocardial fibroelastosis)
See Workup for more detail.
Management
Treatment of DCM is essentially the same as treatment of chronic HF. Some therapeutic interventions treat symptoms, whereas others treat factors that affect survival.
Drug classes used include the following:
-
Angiotensin-converting enzyme (ACE) inhibitors
-
Angiotensin II receptor blockers (ARBs)
-
Beta-blockers
-
Aldosterone antagonists
-
Cardiac glycosides
-
Diuretics
-
Vasodilators
-
Antiarrhythmics
-
Human B-type natriuretic peptide
-
Inotropic agents
-
Neprilysin inhibitor
-
Nitrates
Anticoagulants may be used in selected patients.
Surgical options for patients with disease refractory to medical therapy include the following:
-
Temporary mechanical circulatory support
-
Left ventricular assist devices
-
Cardiac resynchronization therapy (biventricular pacing)
-
Automatic implantable cardioverter-defibrillators
-
Ventricular restoration surgery
-
Heart transplantation
See Treatment and Medication for more detail.
Background
DCM is a progressive disease of heart muscle that is characterized by ventricular chamber enlargement and contractile dysfunction (ie, left ventricular ejection fraction < 40%) (see the image below). [1] The right ventricle may also be dilated and dysfunctional. DCM is the third most common cause of HF and the most frequent reason for heart transplantation. It has an annual incidence of 0.47-0.58 cases per 100,000 and a prevalence of 1 to 4/100,000. [2, 3]
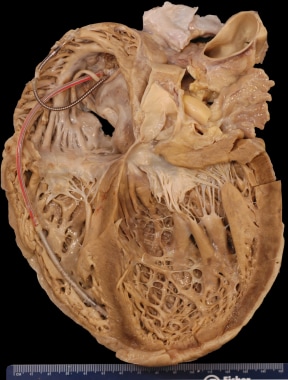 Dilated Cardiomyopathy. Gross heart specimen from a patient with dilated cardiomyopathy who died in end-stage heart failure. Defibrillator leads are in the right heart. The ventricles are dilated with normal ventricular wall thicknesses, imparting an appearance of thin ventricular walls. The ventricles are dilated more than the atria.
Dilated Cardiomyopathy. Gross heart specimen from a patient with dilated cardiomyopathy who died in end-stage heart failure. Defibrillator leads are in the right heart. The ventricles are dilated with normal ventricular wall thicknesses, imparting an appearance of thin ventricular walls. The ventricles are dilated more than the atria.
DCM is one of the three traditional classes of cardiomyopathy, along with hypertrophic and restrictive cardiomyopathy. However, the classification of cardiomyopathies continues to evolve, based on the rapid evolution of molecular genetics as well as the introduction of more recently described diseases.
Multiple causes of DCM exist, one or more of which may be responsible for an individual case of the disease (see Etiology). All alter the normal muscular function of the myocardium, which prompts varying degrees of physiologic compensation for that malfunction.
The degree and time course of malfunction are variable and do not always coincide with a linear expression of symptoms. Persons with cardiomyopathy may have asymptomatic left ventricular (LV) systolic dysfunction, LV diastolic dysfunction, or both. When compensatory mechanisms can no longer maintain cardiac output at normal LV filling pressures, the disease process is expressed with symptoms that collectively compose the disease state known as chronic HF.
Continuing ventricular enlargement and dysfunction generally leads to progressive HF with further decline in LV contractile function. Sequelae include ventricular and supraventricular arrhythmias, conduction system abnormalities, thromboembolism, and sudden death or HF–related death.
Cardiomyopathy is a complex disease process that can affect the heart of a person of any age, but it is especially important as a cause of morbidity and mortality among the world's aging population. It is the most common diagnosis in persons receiving supplemental medical financial assistance via the US Medicare program.
Nonpharmacologic interventions are the basis of HF therapy. Instruction on a sodium diet restricted to 2 g/day is very important and can often eliminate the need for diuretics or permit the use of reduced dosages. Fluid restriction is complementary to a low-sodium diet. Patients may be enrolled in cardiac rehabilitation involving aerobic exercise.
Pathophysiology
DCM is characterized by ventricular chamber enlargement and systolic dysfunction with greater left ventricular (LV) cavity size with little or no wall hypertrophy. Hypertrophy can be judged as the ratio of LV mass to cavity size; this ratio is decreased in persons with DCMs.
The enlargement of the remaining heart chambers is primarily due to LV failure, but it may be secondary to the primary cardiomyopathic process. DCMs are associated with both systolic and diastolic dysfunction. The decrease in systolic function is by far the primary abnormality due to adverse myocardial remodeling that eventually leads to an increase in the end-diastolic and end-systolic volumes. Progressive dilation can lead to significant mitral and tricuspid regurgitation, which may further diminish the cardiac output and increase end-systolic volumes and ventricular wall stress. [1] In turn, this leads to further dilation and myocardial dysfunction.
Early compensation for systolic dysfunction and decreased cardiac output is accomplished by increasing the stroke volume, the heart rate, or both (cardiac output = stroke volume × heart rate), which is also accompanied by an increase in peripheral vascular tone. The increase in peripheral tone helps maintain appropriate blood pressure. Also observed is an increased tissue oxygen extraction rate with a shift in the hemoglobin dissociation curve. In decompensation of systolic HF, several changes in the pressure-volume (P-V) curve are seen. The entire P-V loop shifts to the right with an increase in end-diastolic pressure and end-diastolic volume. Coronary blood flow may also be impaired by hypotension and elevated wall stress, decreasing the perfusion gradient.
The basis for compensation of low cardiac output is explained by the Frank-Starling Law, which states that myocardial force at end-diastole compared with end-systole increases as muscle length increases, thereby generating a greater amount of force as the muscle is stretched. Overstretching, however, leads to failure of the myocardial contractile unit.
Genetic mutations that affect desmin (cytoskeleton), lamin C (nuclear membrane), or myosin (contractile proteins) have been found in association with DCM. [1]
These compensatory mechanisms are blunted in persons with DCMs, as compared with persons with normal LV systolic function. Additionally, these compensatory mechanisms lead to further myocardial injury, dysfunction, and geometric remodeling (concentric or eccentric).
Neurohormonal activation
Decreased cardiac output with resultant reductions in organ perfusion results in neurohormonal activation, including stimulation of the adrenergic nervous system and the renin-angiotensin-aldosterone system (RAAS). Additional factors important to compensatory neurohormonal activation include the release of arginine vasopressin and the secretion of natriuretic peptides. Although these responses are initially compensatory, they ultimately lead to further disease progression.
Alterations in the adrenergic nervous system induce significant increases in circulating levels of dopamine and, especially, norepinephrine. By increasing sympathetic tone and decreasing parasympathetic activity, an increase in cardiac performance (beta-adrenergic receptors) and peripheral tone (alpha-adrenergic receptors) is attempted.
Unfortunately, long-term exposure to high levels of catecholamines leads to down-regulation of receptors in the myocardium and blunting of this response. The response to exercise in reference to circulating catecholamines is also blunted. Theoretically, the increased catecholamine levels observed in cardiomyopathies due to compensation may in themselves be cardiotoxic and lead to further dysfunction. In addition, stimulation of the alpha-adrenergic receptors, which leads to increased peripheral vascular tone, increases the myocardial workload, which can further decrease cardiac output. Circulating norepinephrine levels have been inversely correlated with survival.
Activation of the RAAS is a critical aspect of neurohormonal alterations in persons with chronic HF. Angiotensin II potentiates the effects of norepinephrine by increasing systemic vascular resistance. It also increases the secretion of aldosterone, which facilitates sodium and water retention and may contribute to myocardial fibrosis.
The release of arginine vasopressin from the hypothalamus is controlled by both osmotic (hyponatremia) and nonosmotic stimuli (eg, diuresis, hypotension, angiotensin II). Arginine vasopressin may potentiate the peripheral vascular constriction because of the aforementioned mechanisms. Its actions in the kidneys reduce free-water clearance.
Natriuretic peptide levels are elevated in individuals with DCM. Natriuretic peptides in the human body include atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide. ANP is primarily released by the atria (mostly the right atrium). Right atrial stretch is an important stimulus for its release. The effects of ANP include vasodilation, possible attenuation of cell growth, diuresis, and inhibition of aldosterone. Although BNP was initially identified in brain tissue (hence its name), it is secreted from cardiac ventricles in response to volume or pressure overload. As a result, BNP levels are elevated in patients with congestive HF. BNP causes vasodilation and natriuresis.
Counterregulatory responses to neurohormonal activation involve increased release of prostaglandins and bradykinins. These do not significantly counteract the previously described compensatory mechanisms.
The body's compensatory mechanisms for a failing heart are eventually overwhelmed. Compensation for decreased cardiac output cannot be sustained without inducing further decompensation. The rationale for the most successful medical treatment modalities for cardiomyopathies is therefore based on altering these neurohormonal responses.
Circulating cytokines as mediators of myocardial injury
Tissue necrosis factor-alpha (TNF-alpha) is involved in all forms of cardiac injury. In cardiomyopathies, TNF-alpha has been implicated in the progressive worsening of ventricular function, but the complete mechanism of its actions is poorly understood. Progressive deterioration of LV function and cell death (TNF plays a role in apoptosis) are implicated as some of the mechanisms of TNF-alpha. It also directly depresses myocardial function in a synergistic manner with other interleukins.
Elevated levels of several interleukins have been found in patients with left ventricular dysfunction. Interleukin (IL)-1b has been shown to depress myocardial function. One theory is that elevated levels of IL-2R in patients with class IV congestive HF suggest that T-lymphocytes play a role in advanced HF stages.
IL-6 stimulates hepatic production of C-reactive protein, which serves as a marker of inflammation. IL-6 has also been implicated in the development of myocyte hypertrophy, and elevated levels have been found in patients with congestive HF. IL-6 has been found to correlate with hemodynamic measures in persons with left ventricular dysfunction.
Etiology
Although DCM has many causes, including inherited disease, infections, and toxins, the majority are idiopathic and without an identifiable cause. [1] A systematic approach to define the etiology is essential for determination of the most effective treatment strategy.
Causes of DCM include the following:
-
Heredity or genetics
-
Secondary to other cardiovascular disease: Ischemia, hypertension, valvular disease, tachycardia induced
-
Infectious: Viral, rickettsial, bacterial, fungal, metazoal, protozoal
-
Probable infectious: Whipple disease, Lyme disease
-
Metabolic: Endocrine diseases (eg, hyperthyroidism, hypothyroidism, acromegaly, myxedema, hypoparathyroidism, hyperparathyroidism), diabetes mellitus, electrolyte imbalance (eg, potassium, phosphate, magnesium), pheochromocytoma
-
Rheumatologic/connective tissue disorders: Scleroderma, rheumatoid arthritis, systemic lupus erythematosus
-
Nutritional: Thiamine deficiency (beriberi), protein deficiency, starvation, carnitine deficiency
-
Toxic: Drugs (eg, antineoplastic/anthracycline agents, vascular endothelial growth factor [VEGF] inhibitors), poisons, foods, anesthetic gases, heavy metals, ethanol
-
Collagen vascular disease
-
Infiltrative: Hemochromatosis, amyloidosis, glycogen storage disease
-
Granulomatous: Sarcoidosis, giant cell myocarditis)
-
Physical agents: Extreme temperatures, ionizing radiation, electric shock, nonpenetrating thoracic injury
-
Neuromuscular disorders: Muscular dystrophy (limb-girdle [Erb dystrophy], Duchenne dystrophy, facioscapulohumeral [Landouzy-Dejerine dystrophy]), Friedreich disease, myotonic dystrophy
-
Primary cardiac tumor (myxoma)
-
Senility
-
Peripartum
-
Immunologic: Postvaccination, serum sickness, transplant rejection
-
Stress-induced cardiomyopathy (Takotsubo cardiomyopathy)
In many cases of DCM, the cause remains unexplained. However, some idiopathic cases may result from failure to identify known causes such as infections or toxins. It has been shown that 30-40% of DCM cases have familial inheritance pattern and classified as familial DCM, and efforts have been made to unravel the genetic mechanisms responsible for the DCM in the past couple of decades. [4, 5, 6, 7] The idiopathic category should continue to diminish as more information explaining pathophysiologic mechanisms, specifically genetic-environmental interactions, becomes available.
Toxins are a significant cause. Almost a third of cases may result from severe ethanol abuse (more than 90 grams/day, or 7 to 8 drinks per day) for more than 5 years.
Viral myocarditis
Viral myocarditis is an important entity within the category of infectious cardiomyopathy. Viruses have been implicated in cardiomyopathies as early as the 1950s, when coxsackievirus B was isolated from the myocardium of a newborn baby with a fatal infection. Advances in genetic analysis, such as polymerase chain reaction testing, have aided in the discovery of several viruses that are believed to have roles in viral cardiomyopathies.
Viral infections and viruses associated with myocardial disease may be caused by the following:
-
Coxsackievirus (A and B) [8]
-
Influenza virus (A and B)
-
Adenovirus
-
Echovirus
-
Rabies
-
Hepatitis
-
Yellow fever
-
Lymphocytic choriomeningitis
-
Epidemic hemorrhagic fever
-
Chikungunya fever
-
Dengue fever
-
Cytomegalovirus
-
Epstein-Barr virus
-
Rubeola
-
Rubella
-
Mumps
-
Respiratory syncytial virus
-
Varicella-zoster virus
-
Human immunodeficiency virus
Viral myocarditis can produce variable degrees of illness, ranging from focal disease to diffuse pancarditis involving myocardium, pericardium, and valve structures. Viral myocarditis is usually a self-limited, acute-to-subacute disease of the heart muscle. Symptoms are similar to those of congestive HF and often are subclinical. Many patients experience a flulike prodrome.
Confirming the diagnosis can be difficult because symptoms of HF can occur several months after the initial infection. Patients with viral myocarditis (median age, 42 years) are generally healthy and have no systemic disease.
Acute viral myocarditis can mimic acute myocardial infarction, with patients sometimes presenting in the emergency department with chest pain; nonspecific ECG changes; and abnormal, often highly elevated serum markers such as troponin, creatine kinase, and creatine kinase-MB.
The diagnosis of viral myocarditis is mainly indicated by a compatible history and the absence of other potential etiologies, particularly if it can be supported by acute or convalescent sera. An ECG demonstrates varying degrees of ST-T wave changes reflecting myocarditis and, sometimes, varying degrees of conduction disturbances. Echocardiography is a crucial aid in classifying this disease process, which manifests mostly as a dilated type of cardiomyopathy.
An important diagnostic tool in myocarditis is cardiac MRI, which allows pertinent tissue characterization, specifically, myocardial edema, hyperemia and capillary leak, and necrosis/fibrosis. The classic finding in inflammatory injury is augmented permeability of cell membranes leading to tissue edema, which is detected using T2-weighted imaging. Intertwined with tissue edema is vasodilatation and increased blood tissue delivery to the site of inflammation. As gadolinium is rapidly distributed into the interstitium, using contrast-enhanced fast-spin echo T1-weighted MRI can facilitate myocardial early gadolinium enhancement to assess hyperemia and inflammation. Additionally, late gadolinium enhancement (LGE) can be used to assess necrosis/fibrosis as fibrocytes replace viable tissue in the natural evolution of the disease process. Characteristic distribution of LGE may aid in differentiation of the pathologic processes, such as ischemic versus nonischemic subtypes in which LGE distribution is located in the midwall whereas the subendocardium is involved in ischemia. [9]
Myocarditis is almost always a clinically presumed diagnosis because it is not associated with any pathognomonic sign or specific, acute diagnostic laboratory test result. In the past, percutaneous transvenous right ventricular endomyocardial biopsy has been used, but the Myocarditis Treatment Trial revealed no advantage for immunosuppressive therapy in biopsy-proven myocarditis; therefore, biopsy is not routinely performed in most cases. The diagnostic sensitivity using endomyocardial biopsy is low due to the focal nature of the inflammatory process and to sampling error, leading to increased false negative rates. [10]
Tissue samples are conventionally analyzed by histologic means via light or electron microscopy (Dallas criteria) and modern immunohistochemical methods, but they are not universally assessed by molecular methods of viral genome analysis via polymerase chain reaction (PCR), which would significantly increase the diagnostic potential. Furthermore, on the basis of the combination of absence/scarcity of data on associations of viral loads with clinical outcomes and the uncertain sensitivity of viral genome data, routine testing for viral genome is not recommended outside centers with extensive experience in viral genome analysis. [11]
If a patient is thought to have viral myocarditis, the initial diagnostic strategies should be to evaluate cardiac troponin I or T levels and to perform antimyosin scintigraphy. Positive troponin I or T findings in the absence of myocardial infarction and the proper clinical setting confirm acute myocarditis. Negative antimyosin scintigraphy findings exclude active myocarditis.
The exact mechanism for myocardial injury in viral cardiomyopathy is controversial. Several mechanisms have been proposed based on animal models. Viruses affect myocardiocytes by direct cytotoxic effects and by cell-mediated (T-helper cells) destruction of myofibers. Other mechanisms include disturbances in cellular metabolism, vascular supply of myocytes, and other immunologic mechanisms.
Viral myocarditis may resolve over several months during the treatment of left ventricular systolic dysfunction. However, it can progress to a chronic cardiomyopathy. The main issue in recovery is ventricular size. Reduction of ventricular size is associated with long-term improvement; otherwise, the course of the disease is characterized by progressive dilation.
Because of an immunologic mechanism of myocyte destruction, several trials have investigated the use of immunomodulatory medications. According to Mason et al, the Myocarditis Treatment Trial demonstrated no survival benefit with prednisone plus cyclosporine or azathioprine in patients with viral (lymphocytic) myocarditis. [12]
A randomized study by McNamara et al (Intervention in Myocarditis and Acute Cardiomyopathy [IMAC]) did not show IVIG-treatment–related improvement in left ventricular ejection fraction (LVEF) at 6 and 12 months over placebo. Both groups had similar improvement in LVEF over the study period. [13] In contrast, in a small group of 21 pediatric patients with acute myocarditis, IVIG treatment showed a smaller LV end-diastolic dimension and higher fractional shortening at 12 months. Those treated with IVIG were also more likely to achieve normal LV function and had a higher probability of survival compared to placebo. Although IVIG and immunosuppression are used commonly in myocarditis, a review of studies on immunosuppression in the pediatric population concluded that there was insufficient date for its routine use due to small sample sizes, lack of control group, and differences in medical regimens. [14, 15]
COVID-19 and DCM
COVID-19 has been associated with multiple cardiovascular complications, including cardiomyopathy. Angiotensin-converting enzyme-2 (ACE2) is a functional receptor for the coronavirus 2 (SARS-CoV-2), and it has high expression on respiratory tract and alveolar epithelial cells as well as the myocardium and vascular endothelium. Therefore, in addition to causing pneumonia, COVID-19 can also cause heart and vasculature disease. [16] Myocardial injury ranges between 9.6% and 46.3% in COVID-19 patients. [17] Multiple reports have demonstrated the association between COVID-19 and cardiomyopathy. [18, 19, 20] Although the association of DCM and COVID-19 in adults have not been definitively established, there are several case reports suggesting that COVID-19 can potentially cause DCM in infants and children within age 1 year. [21, 22, 23, 24] Further studies are needed to address the association of DCM and COVID-19.
Familial cardiomyopathy
Familial cardiomyopathy is a term that collectively describes several different inherited forms of HF. Familial DCM is diagnosed in patients with idiopathic cardiomyopathy who have two or more first- or second-degree relatives with the same disease (without defined etiology). Establishing a diagnosis with more-distant affected relatives (third degree and greater) simply requires identifying more family members with the same disease. Thus, molecular genetic testing is recommended for every individual of any age with nonischemic DCM, including those with peripartum or pregnancy-associated cardiomyopathy. [25]
A study by van Spaendonck-Zwarts et al suggested that a subset of peripartum cardiomyopathy is an initial manifestation of familial DCM. This may have important implications for cardiologic screening in such families. [26]
Several forms of familial cardiomyopathy have been described, and theories postulate its association with other causes of cardiomyopathy. These include Barth syndrome, Carvajal syndrome, Duchenne muscular dystrophy, Becker muscular dystrophy, Emery-Dreifuss muscular dystrophy, HFE hemochromatosis, Laing distal myopathy, mitochondrial DCM, and others. [27] Inheritance is mostly autosomal dominant; however, autosomal recessive and sex-linked inheritance have been reported.
Several different genes and chromosomal aberrations have been described in studied families. Mutations include those affecting actin, a cardiac muscle fiber component; titin, a sarcomere structure scaffold; alpha- and beta-myosin heavy chains, which are sarcomeric structural proteins; troponins T, I, or C; dystrophin; and sodium channel mutations.
Anthracycline/doxorubicin-induced cardiomyopathy
Anthracyclines, which are widely used as antineoplastic agents, have a high degree of cardiotoxicity and cause a characteristic form of dose-dependent toxic cardiomyopathy. Both early acute cardiotoxicity and chronic cardiomyopathy have been described with these agents. Anthracyclines can also be associated with acute coronary spasm. The acute toxicity can occur at any point from the onset of exposure to several weeks after drug infusion. Radiation and other agents may potentiate the cardiotoxic effects of anthracyclines.
Cardiac injury occurs even at doses below the empiric limitation of 550 mg/m2. However, whether injury results in clinical congestive HF varies. The development of HF is very rare at total doses less than 450 mg/m2 but is dose dependent.
The history of these patients, in addition to having classic HF symptoms or symptoms of acute myocarditis, involves a previous history of malignancy and treatment with doxorubicin.
Anatomically, these patients' hearts vary from having bilaterally dilated ventricles to being of normal size. The mechanism of myocardial injury is related to degeneration and atrophy of myocardial cells, with loss of myofibrils and cytoplasmic vacuolization. The generation of free radicals by doxorubicin has also been implicated. Progressive deterioration is the norm for this toxic cardiomyopathy. Abnormal myocardial strain analysis by echocardiography precedes changes in LVEF. A peak systolic longitudinal strain reduction by 10-15% during therapy is a good predictor of cardiotoxicity (drop in LVEF of HF). [28]
Prevention is based on limiting dosing after 450 mg/m2 and on serial functional assessments (ie, resting and exercise evaluation of ejection fraction). The drug should be discontinued if the ejection fraction is less than 0.45, if it falls by more than 0.05 from baseline, or if it fails to increase by more than 0.05 with exercise. Dexrazoxane is an iron-chelating agent approved by the FDA to reduce toxicity; however, it increases the risk of severe myelosuppression.
Cardiomyopathy associated with collagen-vascular disease
Several collagen-vascular diseases have been implicated in the development of cardiomyopathies. These include the following:
-
Rheumatoid arthritis
-
Systemic lupus erythematosus
-
Progressive systemic sclerosis
-
Polymyositis
-
HLA-B12–associated cardiac disease
Diagnosis is based on identification of the underlying disease in conjunction with appropriate clinical findings of HF.
Granulomatous cardiomyopathy (sarcoidosis)
Endomyocardial biopsy may be helpful in establishing the diagnosis, especially in sarcoidosis in which the myocardium may be involved. Involvement may be patchy, resulting in a negative biopsy finding. The diagnosis can also be made if some other tissue diagnosis is possible or available in conjunction with the appropriate clinical picture for HF. Cardiac involvement in sarcoidosis reportedly occurs in approximately 20% of cases.
Patients have signs and symptoms of sarcoidosis and congestive HF. Patients rarely present with congestive HF without evidence of systemic sarcoid. Bilateral mediastinal, paratracheal, and/or hilar lymphadenopathy may be evident.
Noncaseating granulomatous infiltration of the myocardium occur as with other organs affected by this disease. Sarcoid granulomas can show a localized distribution within the myocardium. The granulomas particularly affect the conduction system of the heart, left ventricular free wall, septum, papillary muscles, and, infrequently, heart valves. Fibrosis and thinning of the myocardium occurs as a result of the infiltrative process affecting the normal function of the myocardium.
Diagnosis involves finding noncaseating granulomas from cardiac biopsy or other tissues. Often, patients present with conduction disturbances or ventricular arrhythmias. In fact, in patients with normal left ventricular function, these conduction disturbances may be the primary clinical feature.
Treatment of cardiac sarcoidosis with low-dose steroids may be beneficial, especially in patients with progressive disease, conduction defects, or ventricular arrhythmias. The true benefit is unknown because of the lack of placebo-controlled studies. This also holds true for the use of other immunosuppressive agents (eg, chloroquine, hydroxychloroquine, methotrexate) in the treatment of cardiac sarcoidosis.
Giant cell myocarditis
Giant cell myocarditis is a rare, rapidly progressive, and frequently fatal myocarditis. The clinical presentation is typically fulminant HF, ventricular dysrhythmias, and complete heart block. The myocardium is diffusely infiltrated by lymphocytes and multinucleated giant cells. The resulting necrosis and fibrosis leads to ventricular systolic dysfunction and fatal arrhythmias.
The etiology can be secondary to viral infections, autoimmune disorders, and drug hypersensitivity. Inflammatory bowel disease and tumors have also been implicated. Acute cardiac structural findings include wall thickening with normal chamber size that typically dilates with disease progression. Right ventricular dysfunction often follows, which is an independent predictor of death or transplantation. [29]
Right ventricular endomyocardial biopsy (EMB) is used to guide therapy; biopsy has a higher sensitivity (82-85%) than that of other myocarditis counterparts due to its diffuse endocardial involvement for tissue sampling. [30] Early diagnosis is key due to the nature of high mortality of the pathologic process. As such, the 2007 American Heart Association, American College of Cardiology, and European Society of Cardiology (AHA/ACC/ESC) scientific statement recommended EMB as a class IB indication in unexplained new-onset HF of 2 weeks' to 3 months' duration that is associated with a dilated left ventricle, new ventricular arrhythmias, and heart block. [11]
Treatment of giant cell myocarditis is predicated on HF guideline–directed medical therapy plus immunosuppression with cyclosporine and corticosteroids; thus, timely diagnosis via EMB is prudent. [31, 32] Treatment with cyclosporine and corticosteroids is associated with a median transplant-free survival of 12.3 months compared to 3 months without immunosuppression. [33]
The European Study of Epidemiology and Treatment of Cardiac Inflammatory Diseases (ESETCID) showed that there was no benefit in treatment of cytomegalovirus (CMV)-induced myocarditis treated with hyperimmunoglobulin, enterovirus-positive myocarditis treated with interferon alpha, and adenovirus-positive myocarditis treated with IgG and IgM immunoglobulin as compared with placebo. [34]
Hypertensive cardiomyopathy
The classic paradigm of hypertensive heart disease involves concentric left ventricular hypertrophy (LVH) as a mechanism to curtail wall stress, as demonstrated by LaPlace's Law. As the disease progresses (“transition to failure”), the LV dilates and LVEF declines in what is described as a “burned out” LV (eccentric remodeling). This stepwise evolution of the hypertensive heart has been challenged such that progression to concentric versus eccentric remodeling is not set, and that the tendency toward one or the other remains uncertain. However, certain factors such as race (Black persons), female sex, and increased age have a disposition for the development of a concentric response, whereas obesity and lower plasma renin activity are predisposed to eccentric response. Furthermore, the “transition to failure” phase in which the concentric myocardium with intact LVEF progresses to eccentric myocardium with impaired LVEF is uncommon in the absence of myocardial infarction. [35]
The development from asymptomatic LV systolic dysfunction to symptomatic HF (stage B to C) is not completely understood. However, a number of factors appear to govern this transition. First, the transition to decompensation is accelerated by degree of depressed ejection fraction. [36] As cardiac function worsens, compensating mechanisms such as enhanced salt and water retention, increased peripheral vasoconstriction, and increased sympathetic response add further insult and accelerate the development of a decompensated state. The accompanying myocardial remodeling is characterized by fibrosis and LV dilation, with LV geometry taking a less efficient spherical shape; reduced systolic function is considered to play major roles ushering the development of the symptomatic state.
Also noteworthy is the progression of the hypertensive heart with concentric hypertrophy with normal (preserved) ejection fraction (HFpEF) to a symptomatic state. Although the exact mechanism is not well understood, evidence suggests that collagen deposition and titin impact adverse changes in myocardial compliance. [36, 37] Other factors that have been postulated to herald the development of clinical HF include increased mineralocorticoid receptor activation [38] and levels of matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs. [39] Finally, the increased filling pressure is most intertwined with the development of symptomatic HFpEF.
Chagas cardiomyopathy
Chagas disease is caused by Trypanosoma cruzi. The acute presentation is characterized by dyspnea, fever, myalgia, hepatosplenomegaly, and myocarditis. Chronic infection involves the esophagus, colon, and heart. The disease is mainly distributed across Latin America, but Chagas disease may also involve regions in the United States. Cardiac manifestations include biventricular enlargement with dysfunction, apical aneurysm, sinus node dysfunction, and high-degree atrioventricular (AV) block. [40]
Takotsubo (stress) cardiomyopathy
The presentation of Takotsubo cardiomyopathy is similar to acute coronary syndrome (ACS) but in the absence of angiographic evidence of significant coronary artery disease. The classic echocardiographic finding is reversible LV apical ballooning with systolic dysfunction. This condition is triggered by high emotional stress with a preponderance in postmenopausal females. A mild increase in cardiac enzymes with ECG changes, including ST-segment elevation/depression or T-wave changes, may be seen. Postulated pathophysiologic mechanisms include a heightened sympathetic nervous system/catecholaminergic response, coronary vasospasm, and myocarditis. [41]
HIV-associated cardiomyopathy
Cardiac manifestations of infection with HIV include myocarditis, DCM, pericardial effusion, vasculitis, dyslipidemia/insulin resistance secondary to the use protease inhibitors, coronary artery disease, and hypertension secondary to highly active antiretroviral therapy (HAART)-related metabolic syndrome/lipodystrophy. [42] The hazard ratio for death with cardiomyopathy is 4.0. [43]
High-output HF (HOHF)
A high cardiac output is defined as above 8 L/min or a cardiac index beyond 3.9 L/min/m2. The fundamental derangement in HOHF is reduced systemic vascular resistance due to peripheral vasodilation or systemic arteriovenous shunting, with both leading to a decreased mean arterial blood pressure. Consequently, there is a compensatory increase in sympathetic activation, cardiac output, renin-angiotensin-aldosterone system (RAAS), and vasopressin. The result is increased salt/water retention and HF. Conditions that cause increased cardiac output include thyrotoxicosis, beriberi, obesity, anemia, Paget disease, AV malformations and fistulas, and tachycardia syndromes (atrial fibrillation, atrial flutter). Echocardiographic findings of HOHF include compensatory ventricular dilatation and preserved ejection fraction that may deteriorate over time. Mixed venous oxygen saturation is typically over 70%. [44]
Alcoholic cardiomyopathy
Low to moderate levels of alcohol consumption have been shown to have positive cardiovascular benefits; however, excessive, chronic use may lead to myocardial dysfunction. [45] According to the 2013 American College of Cardiology Foundation and American Heart Association (ACCF/AHA) heart failure guidelines, the clinical diagnosis of alcoholic cardiomyopathy is suspected in the presence of biventricular dysfunction and dilation in the setting of excessive alcohol use. [31] The risk is increased in individuals who consume more than 90 g of alcohol daily (approximately 7-8 drinks/day) for longer than 5 years.
The natural evolution of alcoholic cardiomyopathy has not adequately been assessed in light of the available heart failure therapy. A number of studies dating back to the 1970s show rates of overall mortality or the need for transplant ranging from 19% to 73%. Differences were due to different cut-offs in LVEF, variation in the use of beta-blockers/angiotensin converting enzyme (ACE) inhibitors/spironolactone, and the use of implantable cardioverter-defibrillator (ICD)/cardiac resynchronization therapy. [45] Thus, the evolution of alcoholic cardiomyopathy taken in consideration of contemporary therapy, requires further investigation for better understanding. Myocardial recovery has also been described with the cessation of alcohol intake. [46]
Cocaine cardiomyopathy
Cocaine is one of the most abused and addictive psychostimulants, with causal LV systolic depressant rate of 4% to 18%. [31] This drug is a potent sympathomimetic with the potential for devastating cardiovascular consequences, including fatal ventricular arrhythmias, acute myocardial infarction, hypertensive crisis, cerebral vascular accidents, and DCM. [47, 48] Its addictive nature is mediated by its alteration of the dopaminergic activity within the mesocorticolimbic circuitry. Cocaine binds to dopamine, serotonin, and norepinephrine transport proteins, preventing reuptake of these agents into presynaptic neurons, thereby increasing the synaptic presence for enhanced neuroactivity. [49]
Phenotypic characteristics of cocaine cardiomyopathy include chamber dilatation with depressed systolic function, diastolic dysfunction, and LV hypertrophy, particularly in patients with chronic use and secondary hypertension. [48, 50]
Treatment of cocaine cardiomyopathy is similar to that for other DCMs. Note that acute nonselective beta blocker use in the setting of acute cocaine intoxication may result in unopposed alpha-adrenergic receptor stimulation that perpetuates the ongoing insult by increasing coronary vasoconstriction, increasing LV wall stress, and exacerbating hypertensive crisis. [51]
A number of reports have suggested that coronary vascular resistance is significantly increased after administration of beta-blockers, and that animal studies have associated the use of non-vasodilatory beta-blockers to decreased coronary blood flow and higher mortality. [51, 52, 53, 54]
Peripartum cardiomyopathy
Physiologic changes accompanying pregnancy can pose challenges to the cardiovascular system. One of these challenges is peripartum cardiomyopathy (PPCM), which has the potential for significant morbidity and mortality. This condition is characterized by LV systolic dysfunction during the last trimester of pregnancy or the early puerperium period. Cardiomegaly persisting longer than 4-6 months carries a mortality of 50% at 6 years. Subsequent pregnancies in women with cardiomyopathy carries a substantial risk of clinical deterioration, particularly in those who did not recover LV function. In those with recovered LV function, the risk of clinical deterioration is less, but the cardiac dysfunction frequently emerges in the peripartum period. [31, 55] Patients with PPCM should be counseled about the risks that potential pregnancies may have on their health as well as the health of their fetus(es). Genetic forms of PPCM may be at higher risk for clinical nonrecovery. [56]
Cardiovascular changes during pregnancy include expansion of plasma volume, increased cardiac output, and increased activity of the renin-angiotensin-aldosterone system that increases salt and water retention. [55, 56] During labor, there is the potential to overwhelm the cardiovascular system due to the increased cardiac output from tachycardia, catecholamine surges, and deposition of 300-500 mL of blood from the uterus into the maternal circulation. [55]
A number of biomarkers have been studied for the diagnosis and risk stratification of PPCM. The only commercially available marker with adequate efficiency is N-terminal pro b-type natriuretic peptide (NT-proBNP), which is not specific for PPCM but has good sensitivity for heart failure. Other biomarkers of interest include microRNA-146a (MiR-146a), soluble fms-like tyrosine kinase (SFlt1), and cathepsin D (CTSD). [55, 57, 58]
Therapy for PPCM includes standard guideline-directed management, with cautious use of diuretic therapy because placental perfusion may be impaired as well as initiating postdelivery ACE inhibitors due to their teratogenicity. The potential benefit of administering pentoxyifylline or bromocriptine in addition to heart failure medications has been described. [59, 60, 61] Once full myocardial recovery is demonstrated for at least 6 months, a weaning protocol of heart failure therapy can be considered.
Infiltrative cardiomyopathies
Deposition of abnormal substances in patients with infiltrative cardiomyopathies can either increase the LV wall thickness or cause chamber enlargement with the attendant wall thinning and systolic impairment. Increased wall thickness is not necessarily indicative of myocyte hypertrophy; it may be a reflection of the accumulation of intracellular or interstitial substances. Low QRS voltage—despite a “hypertrophic” appearance—is observed more often with interstitial accumulation more than with intracellular accumulation. [62]
Infiltrative cardiomyopathies resembling hypertrophic or hypertensive heart disease
Amyloidosis
The most common type of amyloidosis involving the myocardium results from plasma cell dyscrasias. The extracellular deposition of insoluble amyloid fibrils due to protein misfolding causes predominantly diastolic heart failure, followed by systolic heart failure in advanced stages. [63] Conduction block is also present, as is pericardial involvement, manifested by pericardial effusion. [64] It is diagnosed via EMB or with noninvasive modalities, such as the following:
-
Echocardiography: The echocardiographic appearance of cardiac amyloidosis includes LV and right ventricular wall thickness with a normal chamber size, pericardial effusion, granular myocardial appearance, atrial enlargement, and thickened papillary muscles and valves, with valvular dysfunction if endocardial involvement is present. [62] Two-dimensional speckle tracking demonstrates “apical sparing” of longitudinal strain. The disease involves the four chambers; thus, there is also atrial involvement. Atrial strain reveals impaired atrial systole and diastole and thereby acts as a conduit. The combination of low atrial stroke volume and irregular endocardial deposits due to amyloid deposits leads to a thrombogenic atrium. [65]
-
Cardiac MRI: This imaging modality shows diffuse subendocardium late gadolinium enhancement (LGE) in both ventricles. [66]
-
ECG: The QRS amplitude is decreased.
Fabry disease
Fabry disease is an X-linked recessive lysosomal storage disease that involves alpha galactosidase A deficiency, which leads to accumulation of glycosphingolipid deposition in the myocardium, skin, and kidneys. [67]
Echocardiographic findings of Fabry disease include concentric LV hypertrophy with diastolic dysfunction and normal LVEF and dimension. Although such features may mimic hypertrophic cardiomyopathy, distinguishing characteristics of Fabry disease include the absence of asymmetrical hypertrophy causing LV outflow tract obstruction and a “binary” myocardial appearance due to the increased echogenicity of the subendocardial layer owing to the sphingolipid presence, paralleled by a less echogenic myocardium. [68, 69, 70] Cardiac MRI typically shows focal inferolateral midwall LGE sparing the subendocardium. [71, 72]
Other infiltrative diseases
Other infiltrative diseases that resemble hypertrophic/hypertensive heart disease include Danton disease, Friedreich ataxia, myocardial oxalosis, and mucopolysaccharidoses.
Infiltrative cardiomyopathies resembling DCM
Cardiac sarcoidosis
Cardiac sarcoidosis, a noncaseating granulomatous disease, can involve a number of organs. Cardiac involvement affects the atrioventricular node, causing heart block, as well as the basal septum, papillary muscles, and focal regions in the free wall. [72]
Echocardiographic findings of cardiac sarcoidosis include wall thickening from granulomatous infiltration, with subsequent scarring/thinning that are seen as wall motion abnormalities, LV dilatation, and/or aneurysm. The wall motion abnormalities do not typically correspond to regions subtended by a specific coronary artery. Mitral regurgitation may be seen with papillary muscle involvement. Pulmonary involvement is also common and signs of pulmonary hypertension or right ventricular dysfunction may be present. LGE is patchy and involves the basal and lateral LV walls. [73]
The diagnosis can be made via EMB, although its sensitivity is less than 20%. [74] This is due to the patchy nature of the myocardial involvement.
The use of corticosteroids is the hallmark of treatment and should be started in patients with a high suspicion of cardiac sarcoidosis, even in the presence of a negative biopsy, as early treatment is more effective than later treatment. Unfortunately, therapy does not appear to improve LV volume or function in those with an LVEF below 30%. [75] The presence of concurrent pulmonary sarcoidosis or a depressed LVEF carries a worse prognosis. [76, 77] Immunosuppressants such as methotrexate, azathioprine, and cyclophosphamide may be used in steroid-refractory or steroid-contraindicated cases.
Granulomatosis with polyangiitis (Wegener cardiomyopathy)
Granulomatosis with polyangiitis (formerly Wegener's granulomatosis) is a small- to medium-sized vasculitis that affects the airways, lungs, kidneys, and heart. Cardiac manifestations include pericarditis, supraventricular tachycardias, and heart block. [78] Myocardial involvement/systolic dysfunction has also been described, although not as commonly as the manifestations discussed above. [79, 80] Glucocorticoids and cyclophosphamide are the mainstay of therapy; however, keep in mind that cyclophosphamide itself may cause cardiomyopathy.
Hemochromatosis (iron overload cardiomyopathy)
Iron deposition in the myocardium initially manifests as diastolic dysfunction from a restrictive pathophysiology that progresses to systolic dysfunction. [81] Iron accumulates first in the ventricular myocardium and then the atrial myocardium. [82] As iron itself is proarrhythmic, its involvement in the conduction system may explain the propensity for hemochromatosis toward atrial or ventricular tachyarrhythmias. [83] Iron deposition in the conduction system may cause bradyarrhythmias, warranting placement of pacemakers. [82]
Iron overload is characterized by a transferrin saturation above 55% and a transferrin level over 200 ng/mL for women and over 300 ng/mL for men (on the basis of the 2005 American College of Physicians [ACP] guidelines). [84, 85] However, the level of ferritin in which myocardial deposition is detected is not defined. [81]
As noted earlier, EMB has low sensitivity for hemochromatosis due to the patchy involvement of the myocardium. Echocardiographic findings include ventricular dilatation and restrictive cardiomyopathy. [81] Iron removal may reverse these findings.
Tachycardia-induced cardiomyopathy
Generally, when detected early, tachycardia-induced cardiomyopathy is reversible once treatment of the tachycardia is successful. Common etiologies include chronic untreated atrial fibrillation with rapid ventricular response and frequent (several thousand daily) premature ventricular contractions. Persistent tachycardia is known to lead to myocyte dysfunction and cardiomyopathy. If the tachycardia-induced cardiomyopathy is left untreated, the left ventricular dysfunction can become irreversible. The exact mechanisms by which tachycardia affects cell function are poorly understood. The following are possible mechanisms by which myocyte dysfunction arises from tachycardia:
-
Depletion of energy stores
-
Abnormal calcium channel activity
-
Abnormal subendocardial oxygen delivery secondary to abnormalities in blood flow
-
Reduced responsiveness to beta-adrenergic stimulation
Epidemiology
The true incidence of cardiomyopathies is generally unknown and likely to be underestimated. As with other diseases, authorities depend on reported cases (at necropsy or as a part of clinical disease coding) to define the prevalence and incidence rates. The inconsistency in nomenclature and disease coding classifications for cardiomyopathies has led to collected data that only partially reflect the true incidence of these diseases.
Whether secondary to improved recognition or other factors, the incidence and prevalence of cardiomyopathy appear to be increasing. The reported incidence is 400,000-550,000 cases per year, with a prevalence of 4-5 million people. The estimated prevalence of DCM in adults is 36-40 per 100,000, [86] with a male predominance. [1]
In a 2024 observational study that examined sex differences in the clinical presentation and natural history of DCM using data from 206 females and 398 males with DCM, Owen et al reported a "novel paradox" in females with DCM, observing they have a parodoxical early rise in major HF events despite having less prevalent myocardial fibrosis and a milder phenotype at initial presentation. [87] At 2 years, females had nearly double the risk of cardiovascular mortality or major HF events (8.6%) than males (4.4%). However, there was an attenuation of this effect of sex as a prognostic modifier between 2 and 5 years. [87] Further investigation is needed to understand the underlying mechanism.
Cardiomyopathy is a complex disease process that can affect the heart of a person of any age, and clinical manifestations appear most commonly in the third or fourth decade.
Pediatric population
In children and adolescents, the prevalence of DCM is estimated to be 0.57-1.13 cases per 100,000 people, accounting for about half of all pediatric cardiomyopathies. [88] This incidence is about 10 times lower than that of adults owing to genetic and environmental factors as well as lifestyle habits and comorbidities of adults.
There appears to be a male preponderance (1.32 vs 0.92 in females), and a higher rate of disease occurs in Black patients relative to their White counterparts (1.47 vs 1.06). [88] Most children with DCM are affected within the first 2 years of life, with almost 40% undergoing heart transplantations or dying within 2 years of being diagnosed.
Prognosis
Although some cases of DCM reverse with treatment of the underlying disease, many progress inexorably to HF. With continued decompensation, mechanical circulatory support or heart transplantation may be necessary. Survival is typically poor in the absence of transplantation. [1]
The prognosis for patients with HF depends on several factors, with the etiology of disease being the primary factor. Other factors play important roles in determining prognosis; for example, higher mortality is associated with increased age, male sex, and severe congestive HF. Prognostic indices include the New York Heart Association functional classification.
The Framingham Heart Study found that approximately 50% of patients diagnosed with congestive HF died within 5 years. [89] Patients with severe HF have more than a 50% yearly mortality. Patients with mild HF have significantly better prognoses, especially with optimal medical therapy.
Individuals with a high likelihood of myocardial recovery following appropriate therapy include those with alcohol-induced cardiomyopathy, hypertensive cardiomyopathy, tachycardia-induced cardiomyopathy, Takotsubo cardiomyopathy, or ischemic cardiomyopathy after revascularization.
Cardiopulmonary exercise testing in determining prognosis
An important determinant of prognosis is peak VO2 (oxygen consumption) obtained with cardiopulmonary exercise testing. It is known that an inverse relationship exists between exercise duration and mortality. Because exercise capacity is variable among individuals and reproducibility is not always achieved, exercise testing with respiratory gas analysis provides a standardized method for heart transplant selection. [90] Peak VO2 reflects functional capacity and cardiac reserve. [91] It is a good predictor of mortality, as its decline precedes cardiac decompensation. [92]
Cardiac transplantation appears to have the potential to be deferred in a subset of ambulatory patients with HF. In a study that assessed mortality in 116 patients with chronic HF stratified into three groups, Mancini et al found that peak VO2 was the best predictor for survival, with supporting prognostic information from pulmonary capillary wedge pressure. [91] Group 1 had a peak VO2 below 14 mL/kg/min and was accepted for transplantation; the survival rate at 1 year was 48%. Group 2 had a peak VO2 above 14 mL/kg/min, with patients deemed too well for transplantation; the survival rate at 1 year was 94%, which was comparable to that of their counterparts who underwent transplantation. Group 3 had a peak VO2 below 14 mL/kg/min, along with comorbidities that precluded transplantation; the survival rate at 1 year was 47%.ref70}
The three groups had comparable New York Heart Association functional class, cardiac index, and ejection fraction. Thus, based on these findings on mortality, patients with intact exercise capacity (peak VO2 >14 ml/kg/min) can be medically managed. That is, deferring cardiac transplantation may be safe in ambulatory patients with severe left ventricular dysfunction and a peak exercise VO2 above 14 mL/min/kg. [91] Similarly, Stelken et al showed that a peak VO2 below 50% of the predicted was a strong predictor of 12-month survival in ambulatory patients with HF with an ischemic or dilated etiology. [93]
Ventilatory anaerobic threshold (VAT) is a parameter of cardiopulmonary exercise testing that provides an index of submaximal exercise capacity, independent of patient motivation. It is the point when aerobic metabolism transitions to aerobic plus anaerobic metabolism in which lactate increases. Inability to achieve VAT suggests noncardiovascular limitations of exercise tolerance or poor motivation. [94] In individuals with VAT identified, the reported cardiac event rate was 59% in those with a peak VO2 of 10 mL/kg/min or lower, and 15% in those with a peak VO2 above 18 mL/kg/min. [95] In patients in whom VAT was not detected, the cardiac event rate was 46% in those with peak VO2 of 10 mL/kg/min or below, but for those with a peak VO2 above 10 mL/kg/min, the risk stratification was inconclusive. [95]
Additionally, ventilatory expired gas parameters (VE/VCO2 slope [minute ventilation/carbon dioxide output]) also carry prognostic capability. [96] VE/VCO2 is a ratio relating liters of inspired air to remove 1 L of CO2. A high ratio or slope carries a worse prognosis. Patients with VE/VCO2 slope of 35 or higher had a higher mortality compared to those with a slope below 35 (30% vs 10%, respectively). [96]
-
Dilated Cardiomyopathy. Gross heart specimen from a patient with dilated cardiomyopathy who died in end-stage heart failure. Defibrillator leads are in the right heart. The ventricles are dilated with normal ventricular wall thicknesses, imparting an appearance of thin ventricular walls. The ventricles are dilated more than the atria.
-
Dilated Cardiomyopathy. Heart section from a cardiac explant in a patient with end-stage cardiomyopathy. Note the cardiomyocyte multinucleation. The change is nonspecific and can be seen in heart failure from any cause.
-
Dilated Cardiomyopathy. Heart section from a cardiac explant in a patient with end-stage cardiomyopathy. There is focal interstitial fibrosis. The change is nonspecific and can be seen in heart failure from any cause.
-
Dilated Cardiomyopathy. Heart section from a cardiac explant in a patient with end-stage cardiomyopathy. Note the variation in nuclear size. The change is nonspecific and can be seen in heart failure from any cause.
-
Dilated Cardiomyopathy. Heart section from a cardiac explant in a patient with end-stage cardiomyopathy. Note the intracellular accumulation of amorphous material (basophilic degeneration). The change is nonspecific and can be seen in heart failure from any cause.
Tables

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Treatment
- Approach Considerations
- Blood Pressure Control
- Angiotensin-Converting Enzyme Inhibitors
- Angiotensin Receptor Blockers
- Beta Blockers
- Aldosterone Antagonists
- Cardiac Glycosides
- Diuretics
- Antiarrhythmics
- Vasodilators
- Human B-Type Natriuretic Peptide
- Inotropic Agents
- Anticoagulation
- Left Ventricular Assist Devices
- Cardiac Resynchronization Therapy (Biventricular Pacing)
- Automatic Implantable Cardioverter-Defibrillators
- Heart Transplantation
- Diet and Activity
- Cardiac Rehabilitation
- Investigational Therapy for Heart Failure
- Consultations
- Show All
- Guidelines
- Medication
- Media Gallery
- References