Practice Essentials
Familial renal amyloidosis (FRA) is a group of hereditary disorders in which misfolded proteins—amyloid—accumulate in the kidneys, causing proteinuria and/or hypertension followed by progressive kidney failure. Involvement of other organs (eg, gastrointestinal tract, liver and spleen, heart, skin) varies somewhat depending on the specific genotype.
Susceptibility to FRA is inherited in an autosomal dominant manner (see Presentation/Causes). In nearly all cases, the disease results from mutations in the genes encoding one of the following four plasma proteins:
-
Lysozyme
-
Apolipoprotein AI
-
Apolipoprotein AII
-
Fibrinogen A alpha-chain
However, both penetrance and the clinical phenotype can vary substantially among different families with the same mutation, and even within individual kindreds. Amyloid deposition starts in the first or second decade in some patients, but possibly not until much later in life in others. Consequently, individuals with clinical involvement may present any time from the first decade to old age, although clinical onset most typically occurs in mid-adult life. (See Presentation).
The definitive diagnosis of amyloid accumulation requires histologic confirmation; however, biopsy procedures carry an increased risk of hemorrhage in patients with amyloidosis. (See Workup).
Because organ failure can occur precipitously in organs with extensive amyloid infiltration, treatment of FRA includes scrupulous attention to measures that can reduce the risk of acute kidney injury, as follows:
-
Blood pressure control
-
Salt and water balance
-
Maintenance of circulating volume
-
Prompt treatment of sepsis
-
Avoidance of elective surgery and general anesthesia, unless compelling indications are present
Nevertheless, kidney failure is inevitable in these patients. Renal replacement with hemodialysis or peritoneal dialysis is feasible until a transplant becomes available. In addition to kidney transplantation, liver transplantation may be indicated; liver transplantation is potentially curative in patients with fibrinogen A alpha-chain FRA and, possibly, in some patients with apolipoprotein AI amyloidosis. (See Treatment).
Background
Amyloidosis is a disorder of protein folding in which normally soluble proteins undergo a conformational change and are deposited in the extracellular space in an abnormal fibrillar form. [1] Accumulation of these fibrils causes progressive disruption of the structure and function of tissues and organs, and the systemic (generalized) forms of amyloidosis are frequently fatal. The conditions that underlie amyloid deposition may be either acquired or hereditary, and at least 20 different proteins can form amyloid fibrils in vivo. See the image below.
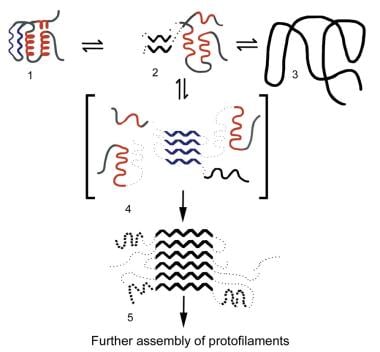 Familial renal amyloidosis. Proposed mechanism for amyloid fibril formation. The drawing depicts a generic amyloid fibril precursor protein (1) in equilibrium with a partially unfolded, molten, globulelike form of the protein (2) and its completely denatured state (3). Autoaggregation through the beta domains initiates fibril formation (4), providing a template for ongoing deposition of precursor proteins and for the development of the stable, mainly beta-sheet, core structure of the fibril (5). The amyloidogenic precursor proteins in patients with familial renal amyloidosis are thought to be less stable than their wild-type counterparts, causing them to populate intermediate, molten, globulelike states more readily.
Familial renal amyloidosis. Proposed mechanism for amyloid fibril formation. The drawing depicts a generic amyloid fibril precursor protein (1) in equilibrium with a partially unfolded, molten, globulelike form of the protein (2) and its completely denatured state (3). Autoaggregation through the beta domains initiates fibril formation (4), providing a template for ongoing deposition of precursor proteins and for the development of the stable, mainly beta-sheet, core structure of the fibril (5). The amyloidogenic precursor proteins in patients with familial renal amyloidosis are thought to be less stable than their wild-type counterparts, causing them to populate intermediate, molten, globulelike states more readily.
Kidney dysfunction is one of the most common presenting features of patients with systemic amyloidosis, and amyloid accumulation is the major pathological finding in approximately 2.5% of all native kidney biopsies. Most such patients have either reactive systemic (AA) amyloidosis or monoclonal immunoglobulin light-chain (AL) amyloidosis, but in the few remaining cases, the disease is hereditary.
The syndrome of familial systemic amyloidosis with predominant nephropathy is inherited in an autosomal dominant manner and was first described in a German family by Ostertag in 1932. [2] Research has shown that almost all patients with familial renal amyloidoses (FRA) are heterozygous for mutations in the genes for lysozyme, apolipoprotein AI, apolipoprotein AII, or fibrinogen A alpha-chain and that the amyloid fibrils in this condition are derived from the respective variant proteins. Both penetrance and the clinical phenotype can vary substantially among different families with the same mutation, and even within individual kindreds.
Pathophysiology
The pathogenesis of amyloid centers around off-pathway folding of the various amyloid fibril precursor proteins. These proteins can exist as two radically different stable structures: the normal soluble form and a highly abnormal fibrillar conformation.
All amyloid fibrils share a common core structure in which the subunit proteins are arranged in a stack of twisted, antiparallel, beta-pleated sheets lying with their long axes perpendicular to the fibril long axis. Proteins that can form amyloid transiently populate partly unfolded intermediate molecular states that expose the beta-sheet domain, enabling them to interact with similar molecules in a highly ordered fashion.
Propagation of the resulting low-molecular-weight aggregates into mature amyloid fibrils is probably a self-perpetuating process that depends only on a sustained supply of the fibril precursor protein. In some cases, the precursors undergo partial proteolytic cleavage; however, whether this occurs before, during, or after the formation of amyloid fibrils remains unknown.
Studies on hereditary amyloidosis have provided unique and valuable insights into the general pathogenesis of amyloid. Most of the variant proteins associated with hereditary amyloidosis differ from their wild-type counterparts by just a single amino acid substitution, although deletions and insertions also occur (see the Table below). [3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21]
Table. Recognized Genotypes and Their Associated Phenotypes in Familial Renal Amyloidosis (Open Table in a new window)
Amyloid Fibril Precursor Protein |
Organs/Tissues Predominantly Affected by Amyloid and Common Clinical Features |
Ethnic Origin of Affected Kindreds |
Lysozyme Ile56Thr |
Renal - Proteinuria and kidney failure Skin - Petechial rashes Liver and spleen - Organomegaly (usually well-preserved function) |
2 British families (possibly related) |
Lysozyme Asp67His |
Renal - Proteinuria and kidney failure GI tract - Bleeding and perforation Liver and spleen - Organomegaly and hepatic hemorrhage Salivary glands – Sicca syndrome |
Single British family |
Lysozyme Try64Arg |
Renal - Proteinuria and kidney failure GI tract - Bleeding and perforation Salivary glands – Sicca syndrome |
Single French family |
Lysozyme Trp82Arg |
Renal - Proteinuria and kidney failure |
Single Chinese family |
Apolipoprotein AI wild type |
Amyloid deposits in aortic atherosclerotic plaques |
20-30% of elderly individuals at autopsy |
Apolipoprotein AI Gly26Arg |
Renal - Proteinuria and kidney failure Gastric mucosa - Peptic ulcers Peripheral nerves - Progressive neuropathy Liver and spleen - Organomegaly (usually well-preserved function) |
Multiple families (mostly of northern European extraction) |
Apolipoprotein AI Trp50Arg |
Renal - Proteinuria and kidney failure Liver and spleen - Organomegaly and liver failure |
Single Ashkenazi family |
Apolipoprotein AI Leu60Arg |
Renal - Proteinuria and kidney failure Liver and spleen - Organomegaly (usually well-preserved function) Cardiac (rarely) - Heart failure |
British and Irish kindreds |
Apolipoprotein AI deletion 60-71 insertion 60-61 |
Liver - Liver failure |
Single Spanish family |
Apolipoprotein AI Leu64Pro |
Renal - Proteinuria and kidney failure Liver and spleen - Organomegaly |
Single Canadian-Italian family |
Apolipoprotein AI deletion 70-72 |
Renal - Proteinuria and kidney failure Liver and spleen - Organomegaly (usually well-preserved function) Retina - Central scotoma |
Single family of British origin |
Apolipoprotein AI Leu75Pro |
Renal - Proteinuria and kidney failure Liver and spleen - Organomegaly |
Italy – Variable penetrance |
Apolipoprotein AI Leu90Pro |
Cardiac - Heart failure Larynx - Dysphonia Skin – Infiltrated yellowish plaques |
Single French family |
Apolipoprotein AI deletion Lys107 |
Aortic intima - Aggressive early-onset ischemic heart disease |
Single Swedish patient at autopsy |
Apolipoprotein AI Arg173Pro |
Cardiac - Heart failure Larynx - Dysphonia Skin - Acanthosis nigricans–like plaques |
British and American families |
Apolipoprotein AI Leu174Ser |
Cardiac - Heart failure |
Single Italian family |
Apolipoprotein AI Ala175Pro |
Larynx - Dysphonia Testicular - Infertility |
Single British family |
Apolipoprotein AILeu178His |
Cardiac - Heart failure Larynx – Dysphonia Skin - Infiltrated plaques Peripheral nerves – Neuropathy |
Single French family |
Apolipoprotein AII Stop78Gly |
Renal - Proteinuria and kidney failure |
American family |
Apolipoprotein AIIStop78Ser |
Renal - Proteinuria and kidney failure |
American family |
Apolipoprotein AIIStop78Arg |
Renal - Proteinuria and kidney failure |
Russian family, Spanish family (different nucleotide substitutions in the two kindreds) |
Fibrinogen A alpha-chain Arg554Leu |
Renal - Proteinuria and kidney failure |
Peruvian, African American and French families |
Fibrinogen A alpha-chain frame shift at codon 522 |
Renal - Proteinuria and kidney failure |
Single French family |
Fibrinogen A alpha-chain frame shift at codon 524 |
Renal - Proteinuria and kidney failure |
Single American family |
Fibrinogen A alpha-chain Glu526Val |
Renal - Proteinuria and kidney failure Late-onset liver (rarely) - Organomegaly and liver failure |
Multiple families (northern European extraction, variable penetrance) |
Fibrinogen A alpha-chain Gly540Val |
Renal - Proteinuria and kidney failure |
Single German family |
Fibrinogen A alpha-chain Indel 517-522 |
Renal - Proteinuria and kidney failure |
Single Korean child |
Investigation of the variant amyloidogenic forms of lysozyme has been exceptionally informative because wild-type lysozyme is not associated with amyloidosis and has been thoroughly characterized. The amyloidogenic mutations give rise to amino acid substitutions that subtly destabilize the native fold so that, under physiological conditions, these variants readily visit partly unfolded states, promoting their spontaneous aggregation into amyloid fibrils.
The whole process of lysozyme amyloid fibril formation can be reversed. A soluble functional variant lysozyme has been recovered in vitro from preparations of isolated ex vivo amyloid fibrils that had been denatured and permitted to refold in the normal conformation. Wild-type apolipoprotein AI is inherently moderately amyloidogenic, and small amyloid deposits derived from it occur in aortic atherosclerotic plaques in 20-30% of middle-aged and elderly individuals.
Amyloid deposits in all different forms of the disease, both in humans and in nonhuman animals, contain the nonfibrillar glycoprotein amyloid P component (AP). AP is identical to and derived from the normal circulating plasma protein, serum amyloid P component (SAP), a member of the pentraxin protein family that includes C-reactive protein.
SAP consists of five identical subunits, each with a molecular mass of 25.462 d, which are noncovalently associated in a pentameric disklike ring. The SAP molecule is highly resistant to proteolysis, and, although not itself a proteinase inhibitor, its reversible binding to amyloid fibrils in vitro protects them against proteolysis. In contrast to its normal rapid clearance from the plasma, SAP persists for very prolonged periods within amyloid deposits. The possibility that SAP may contribute to the pathogenesis and/or persistence of amyloid deposits in vivo has been confirmed in studies on SAP knockout mice. [22]
Amyloid deposits accumulate in the extracellular space, progressively disrupting the normal tissue architecture and consequently impairing organ function. Amyloid deposits can also produce space-occupying effects at both microscopic and macroscopic levels. Although amyloid is inert in the sense that it does not stimulate either a local or systemic inflammatory response, some evidence suggests that the deposits exert cytotoxic effects and possibly promote apoptosis.
Strong clinical impressions exist that suggest the rate of accumulation of amyloid has a major bearing on organ function, which can be preserved for very long periods in the presence of an extensive but stable amyloid load. This may reflect adaptation to gradual amyloid accumulation or may relate to toxic properties of newly formed amyloid material.
Prospective studies with serial SAP scintigraphy, a specific and semiquantitative nuclear medicine technique for imaging amyloid deposits (see Workup/Imaging Studies), have confirmed that amyloid deposits are turned over constantly, albeit at a relatively low and variable rate. Therefore, the course of a particular patient's amyloid disease depends on the relative rates of amyloid deposition versus turnover. Amyloid deposits often regress when the supply of the respective fibril precursor protein is reduced, and, under favorable circumstances, this is accompanied by stabilization or recovery of organ function.
Many questions about amyloid deposition remain unanswered. Why only a small number of unrelated proteins form amyloid in vivo remains unclear, and, as yet, little is known about the genetic or environmental factors that determine individual susceptibility to amyloid or factors that govern its anatomical distribution and clinical effects. Hereditary amyloid deposition starts in the first or second decade in some patients, but possibly not until much later in life in other patients. In addition, the mechanism by which amyloid deposits are cleared and why the rate of this varies so substantially among patients are not understood.
Etiology
Susceptibility to FRA is inherited in an autosomal dominant manner. In nearly all cases, the disease results from mutations in the genes encoding one of the following four plasma proteins:
-
Lysozyme
-
Apolipoprotein AI
-
Apolipoprotein AII
-
Fibrinogen A alpha-chain
In a small number of families, the cause has not yet been determined. Rare cases of renal amyloidosis associated with familial Mediterranean fever have been reported. [23, 24]
Lysozyme
Lysozyme is a ubiquitous bacteriolytic enzyme present in both external secretions and in leukocytes. Lysozyme mutations were identified as a cause of familial amyloidosis when, in 1993, amyloid fibrils in two British families were demonstrated to be derived from the lysozyme variants Asp67His and Ile56Thr, respectively. These represent the least common causes of FRA. The authors have identified a polymorphism encoding lysozyme Thr70Asn, which has an allele frequency of 5% in the British population and which has not been shown to be associated with amyloid deposition.
Apolipoprotein AI
Apolipoprotein AI is a major constituent of high-density lipoprotein (HDL) particles and participates in their central function of reverse cholesterol transport from the periphery to the liver. Approximately half of apolipoprotein AI is synthesized in the liver and half in the small intestine.
Variant forms of apolipoprotein AI are extremely rare in the general population and may be phenotypically silent or may affect lipid metabolism. In 1990, apolipoprotein AI Gly26Arg was identified as a cause of FRA. Since then, 12 other amyloidogenic apolipoprotein AI variants have been discovered. These are mostly other single amino acid substitutions but include deletions and deletion/insertions, not all of which are associated with clinical kidney disease (see Pathophysiology). The amyloid fibril subunit protein has comprised the first 90 or so N-terminal residues of apolipoprotein AI in all cases that have been studied, even when the variant residue(s) has been more distal.
In contrast to lysozyme and fibrinogen A alpha-chain types, wild-type apolipoprotein AI is itself weakly amyloidogenic, and the various amyloidogenic variants are likely to render apolipoprotein AI less stable and/or more susceptible to enzymatic cleavage, promoting an abundance of a fibrillogenic N-terminal fragments.
Another potential mechanism could be reduced lipid binding, thereby increasing the amount of free (and therefore relatively less stable) apolipoprotein AI in the plasma.
Apolipoprotein AII
Apolipoprotein AII is the second major constituent of human HDL particles, accounting for approximately 20% of HDL protein. Like apolipoprotein AI, apolipoprotein AII is synthesized predominantly by the liver and the intestines.
In 2001, apolipoprotein AII stop78Gly was isolated from the amyloid fibrils of a patient who died of kidney failure. [25] Since then, an additional three mutations (encoding two protein variants) have been described in association with hereditary renal amyloidosis (see Pathophysiology).
Unlike serum amyloid A protein (another apolipoprotein and the amyloid precursor in AA amyloidosis) or apolipoprotein AI, in apolipoprotein AII amyloidosis, the protein fibrils are not derived from a cleavage fragment of the native precursor but instead consist of the whole protein plus a 21- amino-acid extension.
Fibrinogen
Fibrinogen is a multimeric 340-kd circulating glycoprotein composed of six peptide chains (two each of alpha, beta, and gamma types), all of which are synthesized in the liver. The alpha chains are the largest and are involved in cross-linking fibrin strands. Numerous alpha-chain variants have been recognized that are either silent or are associated with abnormal hemostasis.
Variant fibrinogen A alpha-chain Arg554Leu was first identified as an amyloid fibril protein in 1993. Since then, five other amyloidogenic mutations have been discovered (see the Table in Pathophysiology). All of these mutations are clustered within the carboxyl terminus of the gene in a relatively small portion of exon 5.
Two of these mutations result in frame shifts that terminate the protein prematurely at codon 548; one is a single nucleotide deletion in the third base of codon 524 and the other is a deletion at codon 522. A single-base transversion, resulting in the substitution of leucine for arginine at codon 554, has been reported in three families of Peruvian-Mexican, African American, and French Caucasian ethnic backgrounds. Residue 554 may be a mutational hot spot because other (nonamyloidogenic) mutations have also been identified at this position.
By far, the most common amyloidogenic variant is fibrinogen A alpha-chain Glu526Val, which has been found in numerous families of Irish, English, German, and Polish origin with FRA.
Genetic information is depicted in the images below.
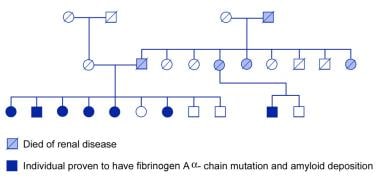 Familial renal amyloidosis. An extended kindred with hereditary amyloidosis associated with fibrinogen A alpha-chain Glu526Val; disease penetrance is high in this particular family.
Familial renal amyloidosis. An extended kindred with hereditary amyloidosis associated with fibrinogen A alpha-chain Glu526Val; disease penetrance is high in this particular family.
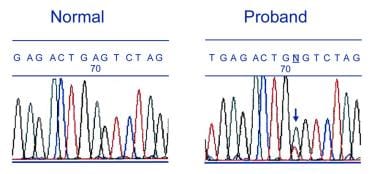 Familial renal amyloidosis. Partial DNA sequence of the gene associated with fibrinogen A alpha-chain Glu526Val in a patient with familial renal amyloidosis, and a sequence from a healthy control. The mutation, which alters codon 526 from glutamic acid to valine, is marked with an arrow.
Familial renal amyloidosis. Partial DNA sequence of the gene associated with fibrinogen A alpha-chain Glu526Val in a patient with familial renal amyloidosis, and a sequence from a healthy control. The mutation, which alters codon 526 from glutamic acid to valine, is marked with an arrow.
Epidemiology
Frequency
No systematic data address the frequency of FRA, but the condition is not as rare as previously thought. The lack of awareness of the condition and the frequent absence of a family history (owing to its variable penetrance) have contributed to substantial underdiagnosis. The incidence of amyloidosis has been estimated at 5 to 13 cases per million population per year; prevalence data are scarce, but one United Kingdom study suggested a rate of about 20 per million inhabitants. [26]
Since the authors introduced routine DNA screening into their investigations of patients with systemic amyloidosis at their facility in the United Kingdom, approximately 5% of patients with presumed AL primary amyloidosis have been diagnosed with hereditary lysozyme, apolipoprotein AI, or fibrinogen A alpha-chain amyloid. The amyloidosis is associated with the fibrinogen A alpha-chain variant Glu526Val in more than 80% of these patients. [27]
Race-, Sex-, and Age-related Demographics
Most patients are of northern European ancestry, but fibrinogen A alpha-chain amyloidosis has been reported in Peruvian-Mexican, Korean, and African-American families, and the authors are presently investigating a northern Indian family with uncharacterized FRA. Cases of fibrinogen A alpha-chain amyloidosis have been reported in two unrelated Chinese individuals. [28]
Gene carriage and the incidence of clinical disease are equal between men and women.
FRA may manifest any time from the first decade to old age but most typically does so in mid-adult life. The age at presentation, like other clinical features, varies among mutations and even within individual kindreds.
Prognosis
Many patients with FRA survive until the seventh decade or older, and most patients survive for at least 10 years after diagnosis. Life expectancy has increased substantially since kidney and liver transplantations have been introduced as treatments for these diseases. Liver transplantation is potentially curative in patients with fibrinogen A alpha-chain FRA and, possibly, in some patients with apolipoprotein AI amyloidosis.
The natural history of familial renal amyloidosis is a relentless gradual progression, leading to kidney and sometimes other organ failure and, eventually, death. Amyloid deposits can ultimately affect many organ systems, but they may be widespread and very extensive without causing symptoms.
The rate of progression and course of disease are extremely variable. Some patients with clinically overt involvement of multiple organs survive for many years or decades. Overall, the prognosis of patients with FRA is much better than that of those with acquired AA and AL amyloidosis.
Histological localization of amyloid deposits determines overall survival in patients with renal amyloidosis. In a study of 35 patients, the glomerulus was the most common and most severely affected renal compartment. Compared with patients without glomerular amyloid deposits, those with severe glomerular amyloidosis advanced more quickly towards end-stage kidney disease and premature death. [29]
Acute kidney injury and chronic kidney disease can occur in the following forms of familial renal amyloidosis (FRA):
-
FRA due to variant lysozyme
-
FRA due to variant apolipoprotein AI
-
FRA due to variant apolipoprotein AII
-
FRA due to variant fibrinogen A alpha-chain
Acute and chronic liver failure can occur in the following forms of FRA:
-
FRA due to variant apolipoprotein AI
-
Potentially, FRA due to variant lysozyme and fibrinogen A alpha-chain (very rarely)
The following complications can occur in these forms of FRA:
-
Restrictive cardiomyopathy - Some apolipoprotein AI and AII variants
-
GI hemorrhage/perforation - Lysozyme FRA
-
Progressive neuropathy - Some patients with apolipoprotein AI Gly26Arg and Leu178His
Patient Education
Patients should be advised to avoid any potential systemic insults such as dehydration, nephrotoxic drugs, and avoidable general anesthetics or surgery. Patients should not only be aware that first-degree relatives each have a 50% chance of carrying the gene but also that disease penetrance is highly variable.
For patient education information, see Amyloidosis.
-
Familial renal amyloidosis. Proposed mechanism for amyloid fibril formation. The drawing depicts a generic amyloid fibril precursor protein (1) in equilibrium with a partially unfolded, molten, globulelike form of the protein (2) and its completely denatured state (3). Autoaggregation through the beta domains initiates fibril formation (4), providing a template for ongoing deposition of precursor proteins and for the development of the stable, mainly beta-sheet, core structure of the fibril (5). The amyloidogenic precursor proteins in patients with familial renal amyloidosis are thought to be less stable than their wild-type counterparts, causing them to populate intermediate, molten, globulelike states more readily.
-
Familial renal amyloidosis. An extended kindred with hereditary amyloidosis associated with fibrinogen A alpha-chain Glu526Val; disease penetrance is high in this particular family.
-
Familial renal amyloidosis. Partial DNA sequence of the gene associated with fibrinogen A alpha-chain Glu526Val in a patient with familial renal amyloidosis, and a sequence from a healthy control. The mutation, which alters codon 526 from glutamic acid to valine, is marked with an arrow.
-
Familial renal amyloidosis. Progression of amyloid deposits in a patient with amyloidosis associated with fibrinogen A alpha-chain Glu526Val. These serial posterior, whole-body, scintigraphic images were obtained following intravenous injection of iodine-123 (123I)–labeled human serum amyloid P component into a 48-year-old man with hereditary amyloidosis associated with fibrinogen A alpha-chain Glu526Val in whom asymptomatic proteinuria had been identified. Both parents were alive and well and older than age 80 years. The scan at diagnosis (left) showed modest abnormal uptake into renal amyloid deposits, which increased at follow-up 3 years later (right). The remainder of the image represents a normal distribution of tracer throughout the blood pool.
-
Familial renal amyloidosis. Regression of amyloidosis associated with fibrinogen A alpha-chain Glu526Val following hepatorenal transplantation. The pictures are serial anterior, whole-body, scintigraphic images obtained following intravenous injection of iodine-123 (123I)–labeled human serum amyloid P component into a patient with amyloidosis associated with fibrinogen A alpha-chain Glu526Val. Prior to hepatorenal transplantation (left), heavy amyloid deposition was present in an enlarged liver and spleen. No amyloid deposits were identified in a follow-up study obtained 42 months after hepatorenal transplantation (right); only a normal distribution of tracer is present throughout the blood pool.
-
Familial renal amyloidosis. Regression of amyloidosis associated with apolipoprotein AI Gly26Arg following hepatorenal transplantation. These serial anterior, whole-body, scintigraphic images were obtained following intravenous injection of iodine-123 (123I)–labeled human serum amyloid P component into a patient with hereditary amyloidosis associated with apolipoprotein AI Gly26Arg. Prior to hepatorenal transplantation (left), heavy amyloid deposition was present in the liver, obscuring the kidneys. Two years after combined hepatorenal transplantation (right), a follow-up scan was normal, showing tracer distributed evenly throughout the background blood pool, including the transplanted organs. Splenic amyloid deposits that were evident initially in posterior scans had regressed at follow-up.
Tables
Amyloid Fibril Precursor Protein |
Organs/Tissues Predominantly Affected by Amyloid and Common Clinical Features |
Ethnic Origin of Affected Kindreds |
Lysozyme Ile56Thr |
Renal - Proteinuria and kidney failure Skin - Petechial rashes Liver and spleen - Organomegaly (usually well-preserved function) |
2 British families (possibly related) |
Lysozyme Asp67His |
Renal - Proteinuria and kidney failure GI tract - Bleeding and perforation Liver and spleen - Organomegaly and hepatic hemorrhage Salivary glands – Sicca syndrome |
Single British family |
Lysozyme Try64Arg |
Renal - Proteinuria and kidney failure GI tract - Bleeding and perforation Salivary glands – Sicca syndrome |
Single French family |
Lysozyme Trp82Arg |
Renal - Proteinuria and kidney failure |
Single Chinese family |
Apolipoprotein AI wild type |
Amyloid deposits in aortic atherosclerotic plaques |
20-30% of elderly individuals at autopsy |
Apolipoprotein AI Gly26Arg |
Renal - Proteinuria and kidney failure Gastric mucosa - Peptic ulcers Peripheral nerves - Progressive neuropathy Liver and spleen - Organomegaly (usually well-preserved function) |
Multiple families (mostly of northern European extraction) |
Apolipoprotein AI Trp50Arg |
Renal - Proteinuria and kidney failure Liver and spleen - Organomegaly and liver failure |
Single Ashkenazi family |
Apolipoprotein AI Leu60Arg |
Renal - Proteinuria and kidney failure Liver and spleen - Organomegaly (usually well-preserved function) Cardiac (rarely) - Heart failure |
British and Irish kindreds |
Apolipoprotein AI deletion 60-71 insertion 60-61 |
Liver - Liver failure |
Single Spanish family |
Apolipoprotein AI Leu64Pro |
Renal - Proteinuria and kidney failure Liver and spleen - Organomegaly |
Single Canadian-Italian family |
Apolipoprotein AI deletion 70-72 |
Renal - Proteinuria and kidney failure Liver and spleen - Organomegaly (usually well-preserved function) Retina - Central scotoma |
Single family of British origin |
Apolipoprotein AI Leu75Pro |
Renal - Proteinuria and kidney failure Liver and spleen - Organomegaly |
Italy – Variable penetrance |
Apolipoprotein AI Leu90Pro |
Cardiac - Heart failure Larynx - Dysphonia Skin – Infiltrated yellowish plaques |
Single French family |
Apolipoprotein AI deletion Lys107 |
Aortic intima - Aggressive early-onset ischemic heart disease |
Single Swedish patient at autopsy |
Apolipoprotein AI Arg173Pro |
Cardiac - Heart failure Larynx - Dysphonia Skin - Acanthosis nigricans–like plaques |
British and American families |
Apolipoprotein AI Leu174Ser |
Cardiac - Heart failure |
Single Italian family |
Apolipoprotein AI Ala175Pro |
Larynx - Dysphonia Testicular - Infertility |
Single British family |
Apolipoprotein AILeu178His |
Cardiac - Heart failure Larynx – Dysphonia Skin - Infiltrated plaques Peripheral nerves – Neuropathy |
Single French family |
Apolipoprotein AII Stop78Gly |
Renal - Proteinuria and kidney failure |
American family |
Apolipoprotein AIIStop78Ser |
Renal - Proteinuria and kidney failure |
American family |
Apolipoprotein AIIStop78Arg |
Renal - Proteinuria and kidney failure |
Russian family, Spanish family (different nucleotide substitutions in the two kindreds) |
Fibrinogen A alpha-chain Arg554Leu |
Renal - Proteinuria and kidney failure |
Peruvian, African American and French families |
Fibrinogen A alpha-chain frame shift at codon 522 |
Renal - Proteinuria and kidney failure |
Single French family |
Fibrinogen A alpha-chain frame shift at codon 524 |
Renal - Proteinuria and kidney failure |
Single American family |
Fibrinogen A alpha-chain Glu526Val |
Renal - Proteinuria and kidney failure Late-onset liver (rarely) - Organomegaly and liver failure |
Multiple families (northern European extraction, variable penetrance) |
Fibrinogen A alpha-chain Gly540Val |
Renal - Proteinuria and kidney failure |
Single German family |
Fibrinogen A alpha-chain Indel 517-522 |
Renal - Proteinuria and kidney failure |
Single Korean child |
