Practice Essentials
Adrenocortical carcinomas (ACs) are rare malignancies, with an annual incidence rate of only 0.7–2 cases per million population in Western countries. [1] ACs can have protean clinical manifestations, including symptoms and signs of hormone excess and nonspecific symptoms as a mass effect of local tumor growth [2] ; however, an increasing number of ACs are identified in asymptomatic patients, as an incidental finding on imaging performed for other reasons (ie, adrenal incidentalomas). [3] Detection of ACs at an early clinical stage is crucial for curative resection; unfortunately, many cases are metastatic at the time of diagnosis , with the most common sites of spread being the local periadrenal tissue, lymph nodes, lungs, liver, and bone. See the image below.
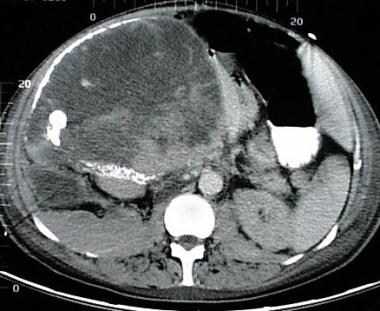 A 68-year-old woman with a large right upper quadrant primary adrenocortical carcinoma with curvilinear calcification. Low-attenuation regions anteriorly are consistent with necrosis.
A 68-year-old woman with a large right upper quadrant primary adrenocortical carcinoma with curvilinear calcification. Low-attenuation regions anteriorly are consistent with necrosis.
Signs and symptoms
Physical examination findings in patients with hormonally active AC include the following:
-
Virilization
-
Cushing syndrome
-
Feminization (rare)
Patients with nonfunctional AC typically present with any of the following:
-
Fever
-
Weight loss
-
Abdominal pain and tenderness
-
Back pain
-
Abdominal fullness
-
Symptoms related to metastases
See Presentation for more detail.
Diagnosis
Laboratory studies
These include the following:
-
Serum glucose
-
Serum cortisol
-
Serum adrenal androgen
-
Urine adrenal hormone
-
Urine vanillylmandelic acid (VMA)
-
Urine homovanillic acid (HVA) levels
Imaging studies
Adrenal computed tomography (CT) scanning and magnetic resonance imaging (MRI) are the imaging studies of choice in AC. The typical case is characterized by a large unilateral adrenal mass with irregular edges. The presence of contiguous adenopathy serves as corroborating evidence.
Histologic examination
Some of the macroscopic features of an AC that suggest malignancy include a weight of more than 500 g, the presence of areas of calcification or necrosis, and a grossly lobulated appearance. Histologic findings also include numerous mitoses, scant cytoplasm, and none of the rosettes observed in neuroblastoma.
See Workup for more detail.
Management
When feasible, total resection remains the modality of choice for the definitive treatment of AC. It also remains the only potentially curative therapeutic modality. While open laparotomy for adrenalectomy represents the standard of care, several reports suggest a role for laparoscopic resection if the adrenal tumor is small and there is no preoperative evidence of metastatic disease.
Medical care in patients with AC, which can be supportive or adjuvant to surgical resection, encompasses the following:
-
Treatment of endocrine excess syndromes
-
Use of mitotane or several multiagent chemotherapy regimens
-
Treatment and prevention of potential complications
-
Strategies for palliative and terminal care issues, including symptom relief and management
See Treatment and Medication for more detail.
Background
Adrenocortical carcinomas (ACs) are uncommon malignancies that can have protean clinical manifestations. A majority of cases are metastatic at the time of diagnosis, with the most common sites of spread being the local periadrenal tissue, lymph nodes, lungs, liver, and bone. ACs are virtually always unilateral. One review suggested that 2-10% of cases may be bilateral at initial diagnosis [4] ; however, this finding has not been replicated. (See Etiology, Pathophysiology, and Workup.)
Adrenocortical masses are common; approximately 2% of the general adult population (7% of those over age 70 years) may have adrenal incidentalomas, biochemically and clinically asymptomatic adrenal masses found incidentally during unrelated imaging investigations, such as abdominal computed tomography (CT) scanning or magnetic resonance imaging (MRI). Only a small number of adrenal incidentalomas (up to 10%) are functional, and only about 2% prove to be ACs. [5] Ectopic adrenocortical tumors are exceedingly rare. [6]
The management of adrenal incidentalomas, therefore, focuses on the following:
- Identifying functional masses and treating them appropriately (including surgical removal)
- Identifying ACs early, with the intent of attempting complete surgical extirpation
- Reassuring patients whose masses are neither functional nor malignant and arranging for follow-up examinations.
Classification of adrenal malignancies
Adrenocortical carcinomas
These include the following:
-
Functional
-
Nonfunctional
-
Well differentiated
-
Intermediate
-
Poorly differentiated to anaplastic
Metastatic adrenal tumors
The most common potential primaries include the following:
-
Lung
-
Breast
-
Melanoma
-
Renal cell carcinoma
-
Extra-adrenal lymphoma
-
Leukemias
-
Pancreatic carcinoma
-
Colonic carcinoma
-
Ovarian carcinoma
Adrenomedullary tumors
These include the following:
-
Malignant pheochromocytoma
-
Ganglioneuroblastoma
-
Neuroblastoma
-
Neuroendocrine carcinoma
Stromal malignancies
These include the following:
-
Neurofibrosarcoma
-
Angiosarcoma
-
Liposarcoma
-
Fibrosarcoma
-
Leiomyosarcoma
-
Myxosarcoma
-
Malignant teratoma
Adrenal malignancies in the setting of familial predisposing syndromes
The associated syndromes include the following:
-
Li-Fraumeni syndrome
-
Familial polyposis coli
-
Gardner syndrome
-
Turcot syndrome
-
Cowden syndrome
-
Beckwith-Wiedemann syndrome (possible)
-
Carney complex (possible)
-
Carney triad
-
Multiple endocrine neoplasia type 1 (MEN1)
Other
These include primary adrenal lymphomas, which can be unilateral or bilateral. Adrenal malignancies can also be classified as composite tumors and mixed tumors.
Functional and nonfunctional tumors
Adrenal tumors are classified in several ways. One popular method, which has great clinical relevance, is to divide them into functional or nonfunctional, depending on the elaboration of adrenocortical hormones (glucocorticoids, mineralocorticoids, androgens, estrogens; rarely, a host of possible peptides).
Nonfunctional variants of AC were previously reported to be far less common than the functional types; older reports suggested that approximately 50-80% of ACs are functional (patients present mainly with Cushing syndrome). Subsequent reports, however, have suggested that nonfunctional ACs may be more common than previously thought.
Sporadic and syndromic tumors
Another classification method is to subdivide ACs into sporadic and syndromic variants. The syndromic variants occur with multiple cancer predisposition syndromes, including Gardner syndrome, Beckwith-Wiedemann syndrome (associated with hemihypertrophy), multiple endocrine neoplasia type 1, the SBLA syndrome (sarcoma, breast, lung, and adrenal carcinoma and other tumors within several kindreds, which have not been clearly associated with localization to a single gene), and Li-Fraumeni syndrome.
Cellular origin
Adrenal tumors can also be classified based on their cellular origin. Included here are primary ACs, primary adrenal lymphomas, soft-tissue sarcomas of the adrenal, malignant pheochromocytomas, and secondary metastatic adrenal tumors (the common primaries of which are tumors of the breast, kidney, lung, and ovary, as well as melanoma, leukemia, and lymphoma). Only the ACs typically are included in discussions of adrenal cancers, and this monograph will be restricted to those. (See Pathophysiology and Etiology.)
Authorities also report rare composite adrenal tumors, which are different histologic variants of the same embryologic origin (eg, coexisting neuroblastoma and malignant pheochromocytoma), and mixed adrenal tumors (typically, mixtures of pheochromocytomas, spindle cell sarcomas, and adrenocortical carcinomas).
Pathophysiology
Endocrine syndromes associated with AC include the following:
-
Cushing syndrome (30%)
-
Virilization and precocious puberty (22%)
-
Feminization (10%)
-
Primary hyperaldosteronism (2.5%)
-
Combined hormone excess (35%)
-
Polycythemia (< 1%)
-
Hypercalcemia (< 1%)
-
Hypoglycemia (< 1%)
-
Adrenal insufficiency (particularly from primary adrenal lymphomas)
-
Non–glucocorticoid-mediated insulin resistance
-
Catecholamine excess due to rare instances of coexisting pheochromocytoma
-
Cachexia (usually preterminal)
Etiology
The exact etiopathogenesis of sporadic AC is unclear, but analysis of syndromic variants of the condition gives some insight. The role of tumor suppressor gene mutations is suggested by their association with Li-Fraumeni syndrome, which is characterized by inactivating germline mutations of the TP53 gene (a vital tumor suppressor gene, or antioncogene) on chromosome 17. This syndrome also is associated with a predisposition to other malignancies, including breast carcinoma, leukemias, osteosarcomas, and soft-tissue sarcomas.
A few reports describe an association between AC and familial adenomatous polyposis, which also is due to an inactivating germline mutation of a tumor suppressor gene (in this case, the adenomatous polyposis coli gene, APC). [7] However, such mutations have not been found in sporadic APC cases.
Hyperplasia
Suggestions have been made that adrenal hyperplasia predisposes patients to develop AC. [8] A few cases of congenital adrenal hyperplasia are associated with functional adrenocortical adenomas but not carcinoma. A few cases of AC are also associated with primary hyperaldosteronism, in which the adrenal tissue has portions showing adrenocortical hyperplasia. However, definitive proof of a sequence in which hyperplasia leads to adenoma, which then leads to carcinoma—similar to a sequence that produces colonic neoplasms—is lacking.
Carney triad
The association of AC with the Carney triad (GI stromal tumor, pulmonary chondromas, extra-adrenal paraganglioma) is far less defined. Since the Carney triad is so rare, there are very few reported cases.
Genetic mechanisms
Potential mechanisms for adrenocortical tumorigenesis include the following:
-
Activation of various proto-oncogenes: Ras, PKC, C myc, C fos, G proteins, and G protein-coupled receptors (eg, for vasoactive intestinal peptide [VIP], gastric-inhibitory peptide [GIP], luteinizing hormone [LH], catecholamines)
-
Inactivation of tumor suppressor genes (antioncogenes): TP53, TP57, TP16, H19, retinoblastoma gene, APC gene, and various deoxyribonucleic acid (DNA) repair ̶ enzyme genes
-
Inhibition of senescence and/or apoptosis: Mutations involving telomerase and/or BCL-2 genes
-
Changes in adrenocortical tissue-specific factors: Mutations involving the genes for StaR, SF-1 (steroidogenic factor), and Dax-1 transcription factor
-
Aberrant expression of receptors to normal adrenocorticotropic agents and ligands: Adrenocorticotropic hormone, angiotensin 2, catecholamines, and endorphins
-
Ectopic expression of receptors on adrenocortical cells to atypical trophic factors and ligands: Cytokines, growth factors, and neurotransmitters
Among the putative pathogenetic mechanisms that may function in concert with each other are alterations in intercellular communication, paracrine and autocrine effects of various growth factors, cytokines elaborated by the tumor cells, and promiscuous expression of various ligand receptors on cell membranes (causing the cells to be in a state of perpetual hyperstimulation). This is presumed to lead to clonal adrenal cellular hyperplasia, autonomous proliferation, tumor formation, and hormone elaboration.
Some molecular studies of adrenocortical tumor cells show in situ mutations of the TP53 and TP57 genes (both antioncogenes) and increased production of insulinlike growth factor 2 (IGF-2). TP53 gene mutations are the most common mutant genes in human cancer. A potential role for this gene in sporadic AC is suggested by the frequent finding of loss of heterozygosity at the 17p13 locus in cases of sporadic AC. Definite germ cell mutations of the TP53 gene have also been demonstrated in more than 90% of children with AC from southern Brazil, which has the highest prevalence of sporadic AC in the world. Amplification of steroidogenic factor-1 expression has also been described in this population.
Another genetic locus of interest is the 11p chromosomal region that may also harbor a tumor suppressor gene and has been implicated in linkage studies in subjects with the Beckwith-Wiedemann syndrome. Loss of heterozygosity at band 11p15 and overexpression of IGF-2, whose gene is carried on this genetic locus, have been described in cases of sporadic AC.
Other studies demonstrate that some of AC cells express menin (the aberrant gene product in patients with multiple endocrine neoplasia type I [MEN1]); in other cases, the hybrid gene is associated with glucocorticoid-responsive aldosteronism (GRA).
Several reports suggest that, while benign adrenal tumors retain expression of the type 2 MHC antigens, these are lost in adrenocortical carcinoma cells. Furthermore, while adrenal adenomas can be monoclonal (43%), polyclonal (28%), or mixed (28%), virtually all ACs are monoclonal.
The fact that the normal adrenal cortex has multiple areas of adrenomedullary cells (often forming large cell nests) and that adrenocortical cells also are scattered in the adrenal medulla suggests a close interaction between the two groups of cells, despite their distinct phylogenetic and embryonic origins. The relevance of the paracrine interactions of these cells in the etiopathogenesis of AC and adrenal tumors as a whole is still being actively investigated.
Epidemiology
AC tumors are uncommon, having an incidence of approximately 0.6-1.67 cases per million persons per year. In southern Brazil, however, the incidence of adrenal tumors is 10-15 times that of the general population, a difference that has been associated with a mutation in the P53 gene.
Sex- and age-related demographics
The female-to-male ratio for ACs is approximately 2.5-3:1. The accumulation of data, especially in international registries, revealed the incidence of adrenal tumors to be higher in female individuals than had previously been thought, particularly in those aged 0-3 years and those over 13 years. Nonfunctional ACs are distributed equally between the sexes.
AC occurs in 2 major peaks: in the first decade of life and again in the fourth to fifth decades. While, functional tumors are more common in children, however, nonfunctional tumors are more common in adults.
Based on data from the International Pediatric Adrenocortical Tumor Registry, the median age at which children develop adrenal carcinomas is 3.2 years; 60% are younger than four years, and 14% are older than 13 years. [9]
Prognosis
AC is relatively rare, accounting for just 0.02-0.2% of all cancer-related deaths. The most important predictive clinical parameters of prognosis are as follows:
-
Disease stage at diagnosis
-
Completeness of resection at surgery
-
Presence or absence of metastasis at the time of diagnosis
Follow-up data from large centers, such as the MD Anderson Cancer Center and the Memorial Sloan-Kettering Cancer Center, suggest a temporal improvement in clinical survival of patients with AC since the late 1980s and early 1990s.
Male patients tend to be older and have a worse overall prognosis than do female patients. Female patients are more likely than male patients to have an associated endocrine syndrome. Although still somewhat controversial, some suggest that children with AC have a better prognosis than do adults; favorable clinical outcome has been reported in 70% or more of pediatric cases. [10]
Detection of tumors at an early clinical stage is crucial for curative resection; total resection offers the only prospect for cure. The estimated overall five-year survival rate for patients with AC is approximately 20-35%. For cases in which total surgical resection is achieved, this rate is estimated to be approximately 32-47%, while in cases in which total surgical extirpation has not been possible, the five-year survival rate is estimated to be 10-30%. [11]
Even after apparently complete surgical resection, however, local or distant relapse occurs in nearly 80% of cases. Documented cases exist of AC recurrence more than 10 years after presumed curative surgery. Recurrent or relapsing AC is usually a bad omen. Although symptoms of hormonal excess can often be medically managed in this setting, cure is virtually unknown.
The presence of distant metastasis generally is another sign of an especially poor outcome. Estimates suggest that as many as 50% of such patients are dead within 12 months of detecting metastatic deposits, regardless of treatment. Indeed, patients with functional AC may have a better prognosis because they present earlier, unlike patients with nonfunctional variants, who invariably present when the tumors are very large or are associated with distant metastasis.
Estrogen receptor (ER)–negative status also confers a worse prognosis in AC. In a study of 17 patients, Shen et al found that one- and five-year survival rates were 86% and 60%, respectively, for patients with ER-positive tumors, versus 38% and 0% for those with ER-negative tumors. [12]
The prognosis for cases of AC occurring in pregnancy is also grim; however, the fetal prognosis in these cases remains excellent.
Patients who show no response to mitotane or who relapse are probably best served by referral to a major cancer center, where they can be enrolled in one of several ongoing combination chemotherapeutic/radiation and/or surgical resection protocols. AC is too uncommon for most tertiary hospitals to have enough expertise to manage these patients adequately.
In 2016, Kim and colleagues published nomograms to predict recurrence-free survival (RFS) and overall survival (OS) after curative resection of adrenocortical carcinoma (ACC). The nomograms were created using a multi-institutional cohort of 148 patients who underwent curative-intent surgery for ACC at 13 major US institutions. [13]
The prediction model for RFS is based on the following 5 independent prognostic factors [13] :
-
Tumor size (< 12 or ≥12 cm)
-
Nodal status (N0, N1, or Nx)
-
T stage (I/II or III/IV)
-
Cortisol-secreting tumor
-
Capsular invasion.
The nomogram to predict OS is based on the following 3 independent prognostic factors [13] :
-
Tumor size (< 12 or ≥12 cm)
-
Nodal status (N0, N1, or Nx)
-
Resection margin (R0 or R1)
Higher total points based on the sum of the assigned number of points for each factor in the nomograms were associated with a worse prognosis. [13]
Complications
Potential complications associated with AC can be subclassified as follows:
-
Local tumor invasion: Including the potential for tumor thrombus formation, which can embolize similar to renal cell carcinoma
-
Hormone excess syndromes (eg, Cushing syndrome, hyperaldosteronism, hirsutism, virilization, hypertension)
-
Paraneoplastic syndromes (eg, cachexia)
-
Local pain in patients with bone metastases
While AC accounts for only approximately 5-10% of cases of Cushing syndrome, approximately 40% of patients with both Cushing syndrome and an adrenal mass also have a malignant tumor. Virtually all feminizing adrenal tumors in men are malignant.
-
A 68-year-old woman with a large right upper quadrant primary adrenocortical carcinoma with curvilinear calcification. Low-attenuation regions anteriorly are consistent with necrosis.
Tables
| Stage | T | N | M |
|---|---|---|---|
| I | T1 | N0 | M0 |
| II | T2 | N0 | MO |
| III | T1-2 |
N1 |
M0 |
T3 |
N0 | M0 | |
| IV | T3 |
N1 |
M0 |
T4 |
Any N |
M0 |
|
Any T |
Any N |
M1 |

What would you like to print?
- Drug Combo for Common Neuroendocrine Tumor: A New Standard?
- Cabozantinib Improves PFS in Advanced Neuroendocrine Tumors After Prior Treatment
- FDA OKs Cabozantinib for Pancreatic Neuroendocrine Tumors
-
 Breast Cancer e-Tumor Boards: Case 5: Metastatic High-Grade Neuroendocrine Carcinoma
Breast Cancer e-Tumor Boards: Case 5: Metastatic High-Grade Neuroendocrine Carcinoma
-
Word Choice and Royals: The Need To Be Clear and Precise
- A Practical Approach to Managing Neuroendocrine Tumors




