Practice Essentials
The most common cause of portal hypertension is cirrhosis. Vascular resistance and blood flow are two important factors in its development. The images below depict esophageal varices, which are responsible for the main complication of portal hypertension, upper gastrointestinal hemorrhage.
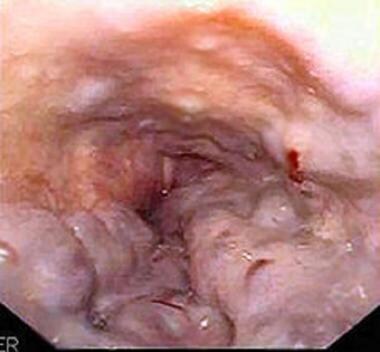 Portal Hypertension. Large esophageal varices with red wale signs seen on endoscopy. Courtesy of Wikimedia Commons.
Portal Hypertension. Large esophageal varices with red wale signs seen on endoscopy. Courtesy of Wikimedia Commons.
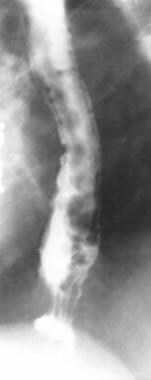 Portal Hypertension. Uphill esophageal varices. Barium swallow demonstrates multiple serpiginous filling defects primarily involving the lower one third of the esophagus with striking prominence around the gastroesophageal junction. The patient had cirrhosis secondary to alcohol abuse.
Portal Hypertension. Uphill esophageal varices. Barium swallow demonstrates multiple serpiginous filling defects primarily involving the lower one third of the esophagus with striking prominence around the gastroesophageal junction. The patient had cirrhosis secondary to alcohol abuse.
Signs and symptoms
Signs and symptoms of liver disease include the following:
-
Weakness, tiredness, and malaise
-
Anorexia, weight loss (common with acute and chronic liver disease)
-
Sudden and massive bleeding, with or without shock on presentation
-
Nausea and vomiting; abdominal discomfort and pain
-
Jaundice or dark urine
-
Edema and abdominal swelling (ascites); splenomegaly
-
Spider angiomas
-
Males: Gynecomastia, testicular atrophy (common with cirrhosis)
-
Pruritus: Usually associated with cholestatic conditions
-
Spontaneous bleeding and easy bruising
-
Symptoms of encephalopathy: Sleep-wake cycle disturbance; intellectual function deterioration, memory loss, and an inability to communicate effectively at any level; personality changes; and, possibly, displays of inappropriate or bizarre behavior
-
Impotence and sexual dysfunction
-
Muscle cramps (common in patients with cirrhosis), muscle wasting
-
Dupuytren contracture (hand deformity)
-
Palmar erythema and leukonychia (white nails): May be present in patients with cirrhosis
-
Asterixis ("flapping tremor," "liver flap")
Complications of portal hypertension may present with the following symptoms:
-
Hematemesis or melena: May indicate gastroesophageal variceal bleeding or bleeding from portal gastropathy
-
Mental status changes: May indicate the presence of portosystemic encephalopathy
-
Increasing abdominal girth: May indicate ascites formation
-
Abdominal pain and fever: May indicate spontaneous bacterial peritonitis, although this disease also presents without symptoms
-
Hematochezia: May indicate bleeding from portal colopathy or enlarged hemorrhoids
Signs of portosystemic collateral formation include the following:
-
Anterior abdominal wall dilated veins: May indicate umbilical epigastric vein shunts
-
Venous pattern on the flanks: May indicate portal-parietal peritoneal shunting
-
Caput medusae (tortuous paraumbilical collateral veins)
-
Rectal hemorrhoids
-
Ascites [1]
-
Paraumbilical hernia
Signs of a hyperdynamic circulatory state include the following:
-
Bounding pulses
-
Warm, well-perfused extremities
-
Arterial hypotension
-
Flow murmur over the pericardium
Other signs of portal hypertension and esophageal varices include the following:
-
Pallor: May suggest active internal bleeding
-
Parotid enlargement: May be related to alcohol abuse and/or malnutrition
-
Cyanosis of the tongue, lips, and peripheries: Due to low oxygen saturation
-
Dyspnea and tachypnea
-
Telangiectasis of the skin, lips, and digits
-
Fetor hepaticus: Occurs in portosystemic encephalopathy of any cause (eg, cirrhosis)
-
Small-sized liver
-
Venous hums: Continuous noises audible in patients with portal hypertension; may be present as a result of rapid, turbulent flow in the collateral veins
-
Tarry stool (digital rectal examination): Suggests upper GI bleeding
-
Hemorrhoids
See Presentation for more detail.
Diagnosis
Laboratory testing
-
Complete blood count
-
Liver disease–associated blood tests (eg, aspartate aminotransferase [AST], alanine aminotransferase [ALT], bilirubin, alkaline phosphatase [ALP])
-
Type and cross-match
-
Coagulation studies (prothrombin time [PT], partial thromboplastin time [PTT], international normalized ratio [INR]): Prolonged INR is suggestive of impaired hepatic synthetic function
-
Albumin: hypoalbuminemia is common (impaired hepatic synthetic function)
-
Blood urea nitrogen, creatinine, and electrolytes
-
Arterial blood gas (ABG) and pH measurements
-
Hepatic and viral hepatitis serologies, particularly hepatitis B and C serologies
Other laboratory tests may include the following:
-
Antinuclear antibody, antimitochondrial antibody, antismooth muscle antibody
-
Iron indices
-
Alpha1-antitrypsin deficiency
-
Ceruloplasmin, 24-hour urinary copper: Consider this test only in individuals aged 3-40 years who have unexplained hepatic, neurologic, or psychiatric disease
Imaging studies
-
Duplex Doppler ultrasonography of the liver and upper abdomen
-
CT scanning and/or MRI: Can be used when ultrasonographic findings are inconclusive
-
Bleeding scan or angiography: Used when bleeding is obscure and the source is unclear
Procedures
-
Liver biopsy and histologic examination
-
Hemodynamic measurement of the hepatic venous pressure gradient (HVPG): A criterion standard for assessment of portal hypertension
-
Upper GI endoscopy (or, esophagogastroduodenoscopy [EGD]): A criterion standard for assessment of varices
See Workup for more detail.
Management
Treatment is directed at the cause of portal hypertension. Gastroesophageal variceal hemorrhage is the most dramatic and lethal complication of portal hypertension; therefore, the focus is on the treatment of variceal hemorrhage. Management of patients with liver cirrhosis and ascites but without hemorrhage includes a low-sodium diet and diuretics.
Emergent treatment
-
Airway, breathing, and circulation evaluation
-
Nasogastric tube placement with hemodynamically significant upper GI bleeding
-
Nothing by mouth; establish two large-bore venous accesses
-
Volume resuscitation, with or without blood product transfusion
-
Portal pressure reduction (ie, anti-secretory agent infusion)
-
Patient transfer to tertiary center with liver transplant service for uncontrolled bleeding from portal hypertension
-
Control and prevention of bleeding from esophageal varices
-
Prevention of complications (eg, prophylactic antibiotics, combination endoscopic/pharmacologic therapy)
-
Administration of vasoconstrictors (eg, octreotide [agent of choice in acute variceal bleeding], vasopressin)
-
Endoscopic therapy (variceal ligation [EVL] [preferred], injection sclerotherapy)
-
Balloon-tube tamponade
-
Percutaneous transhepatic embolization (PTE)
-
Endoscopic administration of cyanoacrylate monomer
-
Transjugular intrahepatic portosystemic shunt (TIPS)
Primary prophylaxis
-
Surveillance
-
Nonselective beta-blockers (eg, propranolol, nadolol, carvedilol)
-
Vasodilators (eg, isosorbide mononitrate [ISMN])
-
Combination pharmacotherapy when a single agent fails
Secondary prophylaxis
-
Nonselective beta-blockers
-
Endoscopic therapy (EVL, treatment of choice; endoscopic sclerotherapy)
-
Combination EVL and pharmacotherapy
Surgery has no role in primary prophylaxis. Consider procedures, such as the following, for the prevention of rebleeding when pharmacologic and/or endoscopic therapy have failed:
-
Portosystemic shunts
-
Devascularization procedures
-
Orthotopic liver transplantation: Treatment of choice for advanced liver disease
See Treatment and Medication for more detail.
Background
Many conditions are associated with portal hypertension, with cirrhosis being the most common cause of this disorder.
Two important factors, vascular resistance and blood flow, play a role in the development of portal hypertension. Ohm law is V = IR, where V is voltage, I is current, and R is resistance. This can be applied to vascular flow; ie, P = FR, where P is the pressure gradient through the portal venous system, F is the volume of blood flowing through the system, and R is the resistance to flow. Changes in either F or R affect the pressure, although in most types of portal hypertension, both of these are altered. (See Anatomy and Etiology and Pathophysiology.)
Normal portal pressure is generally considered to be between 5 and 10 mm Hg. Once the portal pressure rises to 12 mm Hg or greater, complications can arise, such as varices and ascites. Indeed, esophageal varices are responsible for the main complication of portal hypertension, upper GI hemorrhage (see Etiology and Pathophysiology, Prognosis, Presentation, and Workup). (See the images below.)
Portal Hypertension. Uphill esophageal varices. Barium swallow demonstrates multiple serpiginous filling defects primarily involving the lower one third of the esophagus with striking prominence around the gastroesophageal junction. The patient had cirrhosis secondary to alcohol abuse.
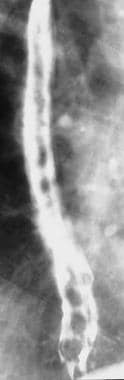 Portal Hypertension. Barium swallow demonstrating esophageal varices involving the entire length of the esophagus. This appearance may be seen in advanced uphill varices or downhill varices secondary to superior vena cava obstruction at or below the level of the azygous vein.
Portal Hypertension. Barium swallow demonstrating esophageal varices involving the entire length of the esophagus. This appearance may be seen in advanced uphill varices or downhill varices secondary to superior vena cava obstruction at or below the level of the azygous vein.
Pathogenesis
The portal vein carries approximately 1500 mL/min of blood from the small and large bowel, the spleen, and the stomach to the liver. [2] Obstruction of portal venous flow, whatever the etiology, results in a rise in portal venous pressure. (See Etiology and Pathophysiology.)
The response to increased venous pressure is the development of collateral circulation that diverts the obstructed blood flow to the systemic veins. These portosystemic collaterals form by the opening and dilatation of preexisting vascular channels connecting the portal venous system and the superior and inferior vena cava. [3, 4, 5] (See the image below.)
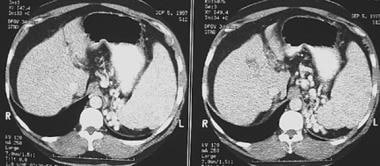 Portal Hypertension. Computed tomography scan showing esophageal varices. Note the extensive collateralization within the abdomen adjacent to the spleen as a result of severe portal hypertension.
Portal Hypertension. Computed tomography scan showing esophageal varices. Note the extensive collateralization within the abdomen adjacent to the spleen as a result of severe portal hypertension.
Although high portal pressure is the main cause of the development of portosystemic collaterals, other factors, such as active angiogenesis, may also be involved. The most important portosystemic anastomoses are the gastroesophageal collaterals, which include esophageal varices. The gastroesophageal collaterals drain into the azygos vein.
Anatomy
The portal vein drains blood from the small and large intestines, stomach, spleen, pancreas, and gallbladder. The superior mesenteric vein and the splenic vein unite behind the neck of the pancreas to form the portal vein. The portal trunk divides into two lobar veins. The right branch drains the cystic vein, and the left branch receives the umbilical and paraumbilical veins that enlarge to form umbilical varices in portal hypertension. The left gastric vein (formerly, gastric coronary vein), which runs along the lesser curvature of the stomach, receives the distal esophageal veins, which also enlarge in portal hypertension. (See the image below.)
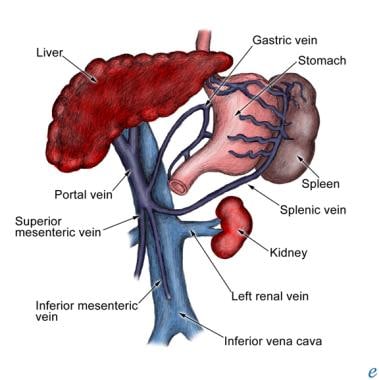
Etiology and Pathophysiology
Increase in vascular resistance
The initial factor in the etiology of portal hypertension is an increase in the vascular resistance to the portal blood flow. The law of Poiseuille, which can be applied to portal vascular resistance, R, states that R = 8hL/pr4, where h is the viscosity of blood, L is the length of the blood vessel, and r is the radius of the blood vessel. The viscosity of the blood is related to the hematocrit. The lengths of the blood vessels in the portal vasculature are relatively constant.
Thus, changes in portal vascular resistance are determined primarily by blood vessel radius. Because portal vascular resistance is indirectly proportional to the fourth power of the vessel radius, small decreases in the vessel radius cause large increases in portal vascular resistance and, therefore, in portal blood pressure (P = F8hL/pr4, where P is portal pressure and F is portal blood flow).
Liver disease that decreases the portal vascular radius produces a dramatic increase in the portal vascular resistance. In cirrhosis, the increase occurs at the hepatic microcirculation (sinusoidal portal hypertension). Increased hepatic vascular resistance in cirrhosis is not only a mechanical consequence of the hepatic architectural disorder; a dynamic component also exists due to the active contraction of myofibroblasts, activated stellate cells, and vascular smooth-muscle cells of the intrahepatic veins.
Endogenous factors and pharmacologic agents that modify the dynamic component include those that increase or decrease hepatic vascular resistance. Factors that increase hepatic vascular resistance include endothelin-1 (ET-1), alpha-adrenergic stimulus, and angiotensin II. Factors that decrease hepatic vascular resistance include nitric oxide (NO), [6] prostacyclin, and vasodilating drugs (eg, organic nitrates, adrenolytics, calcium channel blockers). [4, 7]
Endothelin and nitric oxide
Studies have demonstrated the role of ET-1 and NO in the pathogenesis of portal hypertension and esophageal varices. [4, 7] ET-1 is a powerful vasoconstrictor synthesized by sinusoidal endothelial cells that has been implicated in the increased hepatic vascular resistance of cirrhosis and in the development of liver fibrosis.
NO is a vasodilator substance that is also synthesized by the sinusoidal endothelial cells. In the cirrhotic liver, the production of NO is decreased, and endothelial nitric oxide synthase (eNOS) activity and nitrite production by sinusoidal endothelial cells are reduced. As a result, intrahepatic vasoconstriction occurs in the cirrhotic liver and accounts for approximately 20-30% of the increased intrahepatic resistance. [7, 8, 9] Another major contribution to the increased portal venous pressure is the concomitant splanchnic arteriolar vasodilation causing increased portal venous inflow.
Location of resistance in relation to the liver and sinusoids
Obstruction and increased resistance can occur at three levels in relation to the hepatic sinusoids, as follows (see Table 1, below):
-
Presinusoidal venous block (eg, portal vein thrombosis, schistosomiasis, primary biliary cirrhosis): Characterized by elevated portal venous pressure and a normal wedged hepatic venous pressure (WHVP); these abnormalities cannot be detected by surrogate measurement (WHPV, HVPG), because the measured pressure represents portal pressure in the segment distal to the lesions, which is normal; however, direct measurement of the portal venous pressure will be elevated
-
Postsinusoidal obstruction (eg, right sided heart failure, inferior vena caval obstruction): WHVP is characteristically elevated, whereas the HVPG and FHVP can be either elevated or normal, depending on the site of the obstruction (intrahepatic postsinusoidal vs posthepatic obstruction)
-
Sinusoidal obstruction (eg, cirrhosis): Characterized by HVPG, FHVP, and WHVP, with WHVP being equal to portal venous pressure (because disrupted intersinusoidal communications diminishes compressibility and compliance of the sinusoids, allowing direct transmission of portal pressure to the WHVP)
Table 1. Interpretation of Surrogate Portal Venous Pressure Measurements in the Differential Diagnosis of Portal Hypertension (Open Table in a new window)
Etiology of Portal Hypertension |
WHVP |
FHVP |
HVPG |
|
Prehepatic |
Normal |
Normal |
Normal |
|
Intrahepatic |
Presinusoidal |
Normal |
Normal |
Normal |
Sinusoidal |
Increased |
Increased |
Increased |
|
Postsinusoidal |
Increased |
Normal |
Increased |
|
Posthepatic |
Budd-Chiari syndrome |
N/A |
Hepatic vein cannot be cannulated |
N/A |
Other posthepatic causes |
Increased |
Increased |
Normal |
|
FHVP = free hepatic venous pressure; HVPG = hepatic venous pressure gradient; N/A = not applicable; WHVP = wedged hepatic venous pressure. |
||||
With regard to the liver itself, causes of portal hypertension usually are classified as prehepatic, intrahepatic, and posthepatic.
Prehepatic resistance
Prehepatic causes of increased resistance to flow include the following:
-
Portal vein thrombosis [10]
-
Splenic vein thrombosis
-
Congenital atresia or stenosis of portal vein
-
Extrinsic compression (tumors)
-
Splanchnic arteriovenous fistula
Intrahepatic resistance
Studies of hepatic microcirculation have identified several mechanisms that may explain increased intrahepatic vascular resistance to flow. These mechanisms may be summarized as follows [6] :
-
A reduction of sinusoidal caliber due to hepatocyte enlargement
-
An alteration in the elastic properties of the sinusoidal wall due to collagen deposition in the space of Disse
-
Compression of hepatic venules by regeneration nodules
-
Central vein lesions caused by perivenous fibrosis
-
Veno-occlusive changes
-
Perisinusoidal block by portal inflammation, portal fibrosis, and piecemeal necrosis
More specifically, intrahepatic, predominantly presinusoidal causes of resistance to flow include the following:
-
Schistosomiasis (early stage)
-
Primary biliary cirrhosis (early stage)
-
Idiopathic portal hypertension (early stage) [11]
-
Nodular regenerative hyperplasia: The pathogenesis probably is obliterative venopathy; the presence of nodules that press on the portal system has also been postulated to play a role, although nodularity is present in most cases without clinical evidence of portal hypertension
-
Myeloproliferative diseases: These act via direct infiltration by malignant cells
-
Polycystic disease
-
Hepatic metastasis
-
Granulomatous diseases (sarcoidosis, tuberculosis): Clinical liver dysfunction is rare in sarcoidosis, whereas portal hypertension is an unusual, although well-recognized, manifestation of hepatic sarcoidosis; sarcoid granulomas frequently localize in the portal areas, resulting in injury to the portal veins
Intrahepatic, predominantly sinusoidal causes of resistance include the following:
-
Hepatic cirrhosis
-
Acute alcoholic hepatitis
-
Schistosomiasis (advanced stage)
-
Primary biliary cirrhosis (advanced stage)
-
Idiopathic portal hypertension (advanced stage) [11]
-
Acute and fulminant hepatitis
-
Congenital hepatic fibrosis
-
Peliosis hepatitis
-
Veno-occlusive disease
-
Budd-Chiari syndrome
-
Vitamin A toxicity
-
Sclerosing cholangitis
-
Hepatitis B virus–related and hepatitis C virus–related cirrhosis
-
Wilson disease
-
Hemochromatosis
-
Chronic active hepatitis
With regard to chronic active hepatitis, noncirrhotic portal fibrosis is observed with various toxic injuries, and one of these includes vitamin A toxicity. This probably is due to vascular injury. Excessive doses of vitamin A taken for months or years can lead to chronic hepatic disease. Intake of doses ranging from as small as 3-fold the recommended daily dose continued for several years to doses as high as 20-fold the approved dose for a few months can lead to hepatic disease. The pericellular fibrosis characteristic of vitamin A toxicity may lead to portal hypertension.
Postsinusoidal obstruction syndrome and veno-occlusive disease of the liver are postsinusoidal causes of resistance.
Posthepatic resistance
Posthepatic causes of resistance to flow include the following:
-
Thrombosis of the inferior vena cava (IVC)
-
Right-sided heart failure
-
Constrictive pericarditis
-
Severe tricuspid regurgitation
-
Budd-Chiari syndrome
-
Arterial-portal venous fistula
-
Increased portal blood flow
-
Increased splenic flow
Increase in portal blood flow
The second factor that contributes to the pathogenesis of portal hypertension is an increase in blood flow in the portal veins. This increase is established through splanchnic arteriolar vasodilatation caused by an excessive release of endogenous vasodilators (eg, endothelial, neural, humoral).
The increase in portal blood flow aggravates the increase in portal pressure; the increased flow contributes to the ability of portal hypertension to exist despite the formation of an extensive network of portosystemic collaterals that may divert as much as 80% of the portal blood flow.
Manifestations of splanchnic vasodilatation include increased cardiac output, arterial hypotension, and hypervolemia. This explains the rationale for treating portal hypertension with a low-sodium diet and diuretics to attenuate the hyperkinetic state.
Formation of varices
An elevated pressure difference between systemic and portal circulation (ie, HVPG) directly contributes to the development of varices. [8, 12, 13] HVPG is a surrogate marker of portal pressure gradient and is derived from WHVP corrected (subtracted) with free hepatic venous pressure (FHVP).
The hypertensive portal vein is decompressed by diverting up to 90% of the portal flow through portosystemic collaterals back to the heart, resulting in enlargement of these vessels. These vessels are commonly located at the gastroesophageal junction, where they lie subjacent to the mucosa and present as gastric and esophageal varices. Varices form when the HVPG exceeds 10 mm Hg; they usually do not bleed unless the HVPG exceeds 12 mm Hg (normal HVPG: 1-5 mm Hg). [8, 12, 13, 14]
Gastroesophageal varices have two main inflows. The first is the left gastric vein, and the second is the splenic hilum, through the short gastric veins. The gastroesophageal varices are important because of their propensity to bleed. (See the images below.)
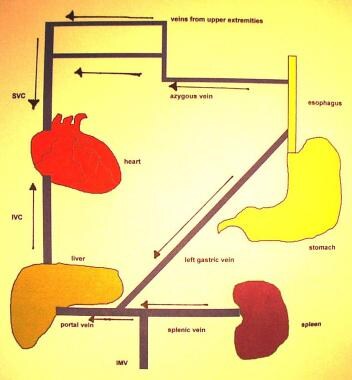 Portal Hypertension. Normal venous flow through the portal and systemic circulation. IMC = inferior mesenteric vein; IVC = inferior vena cava; SVC = superior vena cava.
Portal Hypertension. Normal venous flow through the portal and systemic circulation. IMC = inferior mesenteric vein; IVC = inferior vena cava; SVC = superior vena cava.
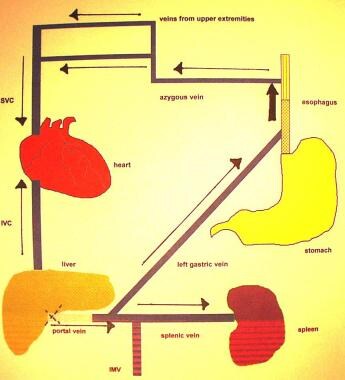 Portal Hypertension. Redirection of flow through the left gastric vein secondary to portal hypertension or portal venous occlusion. Uphill varices develop in the distal one third of the esophagus. IMC = inferior mesenteric vein; IVC = inferior vena cava; SVC = superior vena cava.
Portal Hypertension. Redirection of flow through the left gastric vein secondary to portal hypertension or portal venous occlusion. Uphill varices develop in the distal one third of the esophagus. IMC = inferior mesenteric vein; IVC = inferior vena cava; SVC = superior vena cava.
Mechanisms of variceal hemorrhage
Increased portal pressure contributes to increased varix size and decreased varix wall thickness, thus leading to increased variceal wall tension. Rupture occurs when the wall tension exceeds the elastic limits of the variceal wall. Varices are most superficial at the gastroesophageal junction and have the thinnest wall in that region; thus, variceal hemorrhage invariably occurs in that area. [14]
The following are risk factors for variceal hemorrhage [8, 12, 15] :
-
Variceal size: The larger the varix, the higher the risk of rupture and bleeding; however, patients may bleed from small varices too
-
The presence of endoscopic red color signs (eg, red wale markings, cherry red spots)
-
Child B or C classification, especially the presence of ascites, increases the risk of hemorrhage
-
Active alcohol intake in patients with chronic, alcohol-related liver diseases
-
Bacterial infection: A well-documented association exists between variceal hemorrhage and bacterial infections, and this may represent a causal relationship
Note that bacterial infection could also trigger variceal bleeding through a number of mechanisms, including the following:
-
The release of endotoxin into the systemic circulation
-
Worsening of hemostasis
-
Vasoconstriction induced by the contraction of stellate cells
Epidemiology
Population-based prevalence data for portal hypertension in the United States are not available, but portal hypertension is a frequent manifestation of liver cirrhosis. According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA), liver cirrhosis accounted for almost 30,000 deaths in the United States in 2007, making it the 12th leading cause of US deaths. [18]
The international incidence of portal hypertension is also not known, although it is probably similar to that of the US, with differences primarily in the causes. In Western countries, alcoholic and viral cirrhosis are the leading causes of portal hypertension and esophageal varices; 30% of patients with compensated cirrhosis and 60-70% of patients with decompensated cirrhosis have gastroesophageal varices at the time of diagnosis. [8, 12] The frequency of gastroesophageal varices directly correlates with the severity of the liver disease from 40% in Child class A to 85% in Child class C. [8, 12]
The de novo rate of development of esophageal varices in US patients with chronic liver disease is approximately 8% per year for the first 2 years and 30% by the sixth year. The risk of bleeding from esophageal varices is 30% in the first year after identification. Bleeding from esophageal varices accounts for approximately 10% of episodes of upper gastrointestinal bleeding.
Hepatitis B is endemic in the Far East and Southeast Asia, particularly, as well as in South America, North Africa, Egypt, and other countries in the Middle East. Schistosomiasis is an important cause of portal hypertension in Egypt, Sudan, southern and sub-Saharan Africa, Southeast Asia, Caribbean, and South America. Nonalcoholic steatohepatitis (NASH) is becoming a major cause of liver cirrhosis in the United States as hepatitis C is becoming a major cause of liver cirrhosis worldwide.
Sex- and age-related demographics
Liver disease demonstrates a sex predilection, with males making up more than 60% of patients with chronic liver disease and cirrhosis. [19]
In general, alcoholic liver disease and viral hepatitis are the most common causes of esophageal varices in both sexes. However, veno-occlusive diseases and primary biliary cholangitis are more common in females; and in females with esophageal varices, alcoholic liver disease, viral hepatitis, veno-occlusive disease, and primary biliary cholangitis are usually responsible. In males with esophageal varices, alcoholic liver disease and viral hepatitis are usually the cause.
Portal vein thrombosis and secondary biliary cirrhosis are the most common causes of esophageal varices in children. Cirrhosis is the most common cause of esophageal varices in adults.
Prognosis
Portal hypertension is a signficant cause of morbidity and mortality in patients with cirrhosis, [20] particularly among those with advanced liver disease. [21] In a multicenter retrospective study (2017-2019) comprising data from 3836 patients (2% with mild liver disease, 3% with moderate-severe liver disease) admitted to intensive care units (ICUs), White et al found an increased risk for 90-day case fatality in those with moderate-severe chronic liver disease (CLD) (41%) following ICU admission compared to 25% for those with mild CLD and 17% for individuals without CLD. [21] They noted that moderate-severe CLD was an independent factor associated with higher 90-day case fatality, but not mild CLD.
Patients with severe and persistent upper gastrointestinal hemorrhage (ie, requiring transfusions of more than 5 units of packed red blood cells) have a higher morbidity and mortality rate.
Variceal hemorrhage is the most common complication associated with portal hypertension. Almost 90% of patients with cirrhosis develop varices, and approximately 30% of varices bleed. The estimated mortality rate for the first episode of variceal hemorrhage is 30-50%.
Patients with a known diagnosis of esophageal varices have a 30% chance of variceal bleeding within the first year after the diagnosis. [12] The mortality rate of the bleeding episode depends on the severity of the underlying liver disease. [12]
Patients who have had one episode of bleeding from esophageal varices have a 60-80% chance of rebleeding within 1 year after the initial episode; approximately one third of further bleeding episodes are fatal. [8, 12, 22] The risk of death is maximal during the first few days after the bleeding episode and decreases slowly over the first 6 weeks. However, despite improvements in therapy, the mortality rate at 6 weeks is remains greater than 20%; this rate is higher when surgical intervention is needed. [8]
Associated abnormalities in the renal, pulmonary, cardiovascular, and immune systems of patients with esophageal varices contribute to 20-65% of deaths in these individuals. Complications associated with portal hypertension and GI bleeding include the following:
-
Hepatic encephalopathy
-
Bronchial aspiration, aspiration pneumonia
-
Renal failure
-
Systemic infections, sepsis
-
Spontaneous bacterial peritonitis
-
Ascites
-
Hepatorenal syndrome
-
Bacteremia and/or endotoxemia
-
Vascular collapse
-
Cardiomyopathy
-
Arrhythmias
-
Hypotension
-
Portal hypertensive gastropathy: This is a common complication of cirrhosis and portal hypertension, but significant bleeding from this source is relatively uncommon
Other complications include those related to blood transfusion(s) and/or those related to the therapeutic procedures used in the management of bleeding varices.
In a retrospective study (2002-2014) of 80 patients with portopulmonary hypertension, Mayo Clinic investigators noted that intrapulmonary vascular dilatations (IPVDs) were common and associated with reduced survival. [23] The presence of IPVDs was detected by agitated saline contrast-enhanced transthoracic echocardiography (cTTE), traditionally more frequently associated with hepatopulmonary syndrome than with portopulmonary hypertension. [23]
Prognosis in patients with esophageal varices
Patients with a hepatic venous pressure gradient (HVPG) of 20 mm Hg measured 24 hours after the onset of bleeding esophageal varices have a higher 1-year mortality rate. [12] Other factors that can affect the prognosis of patients with esophageal varices include the following:
-
Rebleeding
-
Child classification: Especially the presence of ascites
-
Active alcohol intake in patients with chronic, alcohol-related liver diseases
-
Occurrence of complications
Several factors are known to influence the prognosis of esophageal bleeding. These include the following:
-
The natural course of the disease causing portal hypertension
-
The severity of the portal hypertension
-
The location and number of the bleeding varices
-
The functional status of the liver and the severity of the liver disease: Early rebleeding, within 5 days of admission; occurred in 21% of patients classified as Child-Pugh grade A, 40% of patients classified as grade B, and 63% of patients classified as grade C
-
Presence of associated systemic disorders
-
Continued alcohol abuse
-
Response to emergency treatment
Patient Education
Educate patients about the benefits and disadvantages of available treatment options.
Alcohol intake should strongly be discouraged, especially in patients with alcoholic cirrhosis. Available resources for alcohol rehabilitation should be provided, along with any prophylaxis for alcohol withdrawal symptoms, when indicated.
Unless contraindicated, all patients with esophageal varices should take beta-blockers to reduce the risk of bleeding. Patients should also be educated about the adverse effects of beta-blockers and the possible risks of their abrupt discontinuation.
Advise patients who have ascites of the risk of spontaneous bacterial peritonitis during an episode of acute variceal bleeding.
-
Portal Hypertension. Large esophageal varices with red wale signs seen on endoscopy. Courtesy of Wikimedia Commons.
-
Portal Hypertension. Uphill esophageal varices. Barium swallow demonstrates multiple serpiginous filling defects primarily involving the lower one third of the esophagus with striking prominence around the gastroesophageal junction. The patient had cirrhosis secondary to alcohol abuse.
-
Portal Hypertension. Barium swallow demonstrating esophageal varices involving the entire length of the esophagus. This appearance may be seen in advanced uphill varices or downhill varices secondary to superior vena cava obstruction at or below the level of the azygous vein.
-
Portal Hypertension. Computed tomography scan showing esophageal varices. Note the extensive collateralization within the abdomen adjacent to the spleen as a result of severe portal hypertension.
-
Portal Hypertension. Normal venous flow through the portal and systemic circulation. IMC = inferior mesenteric vein; IVC = inferior vena cava; SVC = superior vena cava.
-
Portal Hypertension. Redirection of flow through the left gastric vein secondary to portal hypertension or portal venous occlusion. Uphill varices develop in the distal one third of the esophagus. IMC = inferior mesenteric vein; IVC = inferior vena cava; SVC = superior vena cava.
-
Portal Hypertension. Portal vein and associated anatomy.
-
Portal Hypertension. Power Doppler sonogram through the spleen shows varices at the hilum of an enlarged spleen. The final diagnosis was hepatitis C cirrhosis, hepatocellular carcinoma of the left hepatic lobe (which had ruptured into the peritoneum), and portoarterial fistula (which had developed inside the ruptured tumor, giving rise to severe portal hypertension).
-
Portal Hypertension. Duplex spectral Doppler sonogram of the portal vein (same patient as in the previous image) shows a bidirectional flow within the vein. The final diagnosis was hepatitis C cirrhosis, hepatocellular carcinoma of the left hepatic lobe (which had ruptured into the peritoneum), and portoarterial fistula (which had developed inside the ruptured tumor, giving rise to severe portal hypertension).
-
Portal Hypertension. Digital subtraction selective common hepatic artery angiogram shows immediate filling of the portal venous radicles in the left lobe of the liver (straight arrow) and early filling of portal vein (curved arrow), suggestive of hepatic arterial-portal vein fistula. The final diagnosis was hepatitis C cirrhosis, hepatocellular carcinoma of the left hepatic lobe (which had ruptured into the peritoneum), and portoarterial fistula (which had developed inside the ruptured tumor, giving rise to severe portal hypertension).
-
Portal Hypertension. Delayed venous phase of a selective common hepatic angiogram (same patient as in the previous image) shows the portal vein (P), with filling of the left gastric vein caused by retrograde flow feeding gastric and lower esophageal varices (arrows). Retrograde flow in enlarged umbilical veins also is seen. The final diagnosis was hepatitis C cirrhosis, hepatocellular carcinoma of the left hepatic lobe (which had ruptured into the peritoneum), and portoarterial fistula (which had developed inside the ruptured tumor, giving rise to severe portal hypertension).
-
Portal Hypertension. Digital subtraction venous phase of a superior mesenteric artery angiogram (same patient as in the previous 2 images) shows retrograde flow into the left gastric vein (curved arrow) and the inferior mesenteric vein (straight arrow). Note the flow defect of the distal portal vein caused by retrograde flow (open arrowhead). The final diagnosis was hepatitis C cirrhosis, hepatocellular carcinoma of the left hepatic lobe (which had ruptured into the peritoneum), and portoarterial fistula (which had developed inside the ruptured tumor, giving rise to severe portal hypertension).
-
Portal Hypertension. This video, captured via esophagoscopy, shows band ligation of esophageal varices. Video courtesy of Dan C Cohen, MD, and Dawn Sears, MD, Division of Gastroenterology, Scott & White Healthcare.
Tables
Etiology of Portal Hypertension |
WHVP |
FHVP |
HVPG |
|
Prehepatic |
Normal |
Normal |
Normal |
|
Intrahepatic |
Presinusoidal |
Normal |
Normal |
Normal |
Sinusoidal |
Increased |
Increased |
Increased |
|
Postsinusoidal |
Increased |
Normal |
Increased |
|
Posthepatic |
Budd-Chiari syndrome |
N/A |
Hepatic vein cannot be cannulated |
N/A |
Other posthepatic causes |
Increased |
Increased |
Normal |
|
FHVP = free hepatic venous pressure; HVPG = hepatic venous pressure gradient; N/A = not applicable; WHVP = wedged hepatic venous pressure. |
||||

What would you like to print?
- Hypertension Apps Make a Small Difference
- FDA Approves Triple Rx for Hypertension
- Epistaxis and Hypertension: Reviewing a Complex Relationship
-
 Jun 27, 2025 This Week in Cardiology Podcast
Jun 27, 2025 This Week in Cardiology Podcast
-
European Alliance of Associations for Rheumatology (EULAR) 2025 Annual Meeting
- Influence of Obesity on Invasive Hemodynamics and Prognosis in Patients With Heart Failure





