Practice Essentials
Membranoproliferative glomerulonephritis (MPGN) is an uncommon cause of chronic nephritis that occurs primarily in children and young adults. These patients have a pattern of glomerular injury with the following three characteristic histopathologic findings:
-
Proliferation of mesangial and endothelial cells and expansion of the mesangial matrix (see the first image below)
-
Thickening of the peripheral capillary walls by subendothelial immune deposits and/or intramembranous dense deposits (see the second image below)
-
Mesangial interposition into the capillary wall, giving rise to a double-contour or tram-track appearance on light microscopy (see the third image below)
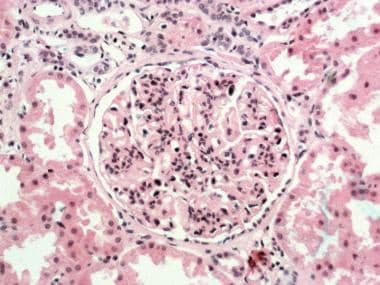 Glomerulus with lobular accentuation from increased mesangial cellularity. A segmental increase occurs in the mesangial matrix, and the peripheral capillary walls are thickened (hematoxylin and eosin stained section; original magnification × 250). Courtesy of John A. Minielly, MD.
Glomerulus with lobular accentuation from increased mesangial cellularity. A segmental increase occurs in the mesangial matrix, and the peripheral capillary walls are thickened (hematoxylin and eosin stained section; original magnification × 250). Courtesy of John A. Minielly, MD.
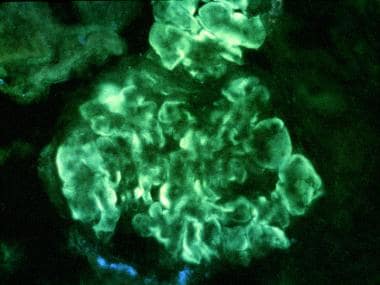 Immunofluorescent stained section. Intense, peripheral, glomerular, capillary loop deposition of immunoglobulin G (IgG) in an interrupted linear pattern corresponding to extensive subendothelial immune deposits (original magnification × 400). Courtesy of John A. Minielly, MD.
Immunofluorescent stained section. Intense, peripheral, glomerular, capillary loop deposition of immunoglobulin G (IgG) in an interrupted linear pattern corresponding to extensive subendothelial immune deposits (original magnification × 400). Courtesy of John A. Minielly, MD.
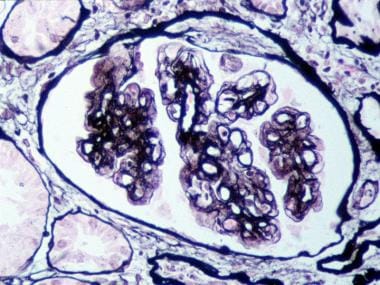 Glomerulus with mesangial interposition producing a double contouring of basement membranes, which, in areas, appear to surround subendothelial deposits (Jones silver methenamine–stained section; original magnification × 400). Courtesy of John A. Minielly, MD.
Glomerulus with mesangial interposition producing a double contouring of basement membranes, which, in areas, appear to surround subendothelial deposits (Jones silver methenamine–stained section; original magnification × 400). Courtesy of John A. Minielly, MD.
MPGN may be idiopathic or secondary in etiology. [1] The secondary types are more common than the idiopathic types and are diagnosed by carefully reviewing clinical features, laboratory data, and renal histopathology. Historically, primary (idiopathic) MPGN was subdivided into types I, II, and III based on ultrastructural appearance on electron microscopy, as follows:
-
Type I – Subendothelial deposits
-
Type II – Dense deposits in the glomerular basement membrane
-
Type III – Subepithelial and subendothelial
Pathophysiology
The normal complement system consists of the classic and alternative pathways. The classic pathway is activated by the interaction of C1 with an antigen-antibody complex. This interaction results in the formation of C4b2a, which is the classic pathway C3b convertase. The alternative pathway utilizes C3 and factors B and D to form the alternative pathway convertase C3b,Bb.
Small amounts of C3b are constantly being formed in the circulation, and are inactivated by factors H and I. The binding of C3b to a foreign antigen decreases its affinity for factor H and allows for the formation of increasing amounts of the alternate pathway convertase. The classic and alternate pathway convertases cause C3 activation, forming C3a and C3b. C3b is an opsonin itself, and C3 convertase facilitates the activation of the terminal pathway and the formation of the membrane attack complex C5b-9.
Hypocomplementemia in MPGN
Hypocomplementemia is a characteristic finding with all types of membranoproliferative glomerulonephritis (MPGN). Low C3 levels are present in approximately 75% of patients with this condition. Although hypocomplementemia bears no relation to the clinical course or prognosis of MPGN, it plays a role in initiating glomerular inflammation and injury. Hypocomplementemia results from increased catabolism and decreased C3 synthesis. The decreased C3 synthesis is likely caused by the negative feedback by C3 breakdown products.
Three nephritic antibodies are described in MPGN that play a role in the development of hypocomplementemia [6, 7] : (
-
Nephritic factor of the classic pathway (NFc or C4NeF)
-
Nephritic factor of the amplification loop (NFa or C3NeF)
-
Nephritic factor of the terminal pathway (NFt)
The reason for the generation of nephritic antibodies is not known. These autoantibodies are not specific for MPGN and are also seen in poststreptococcal and lupus glomerulonephritis. NFc stabilizes the classic pathway C3 convertase C4b,2a. This nephritic factor does not cause C3 conversion unless C4b,2a production is ongoing. NFa (C3NEF) is an autoantibody to C3b,Bb. The binding of NFa to C3b,Bb stabilizes the complex, preventing degradation by its normal inactivators, resulting in complement activation and chronic consumption of C3.
NFt stabilizes the alternative pathway properdin-dependent C3/C5 convertase (C3Bb2,Bb,P) and leads to C3 activation and consumption. The consumption of C3 caused by NFt is much slower than that caused by NFa. NFt also activates the terminal complement components forming C5b-C9, the membrane attack complex.
Immune complex–mediated glomerulonephritis (ICGN)
In all patients with ICGN, deposits of complement and immunoglobulin are found in the mesangium and subendothelial spaces. These trigger release of cytokines and chemokines, causing an influx of inflammatory cells and leading to mesangial and endothelial cell proliferation. Most patients with circulating immune complexes do not develop MPGN and ICGN can certainly be seen without circulating immune complexes; thus, additional pathogenic factors (eg, nature of the antigen, size of complexes, type and charge on antibodies, local glomerular factors) must play a role. Common etiologies include infections, autoimmune disease, or monoclonal gammopathies.
However, many patients with monoclonal deposits on immunofluorescence or ultrastructural evidence on electron microscopy do not have an identifiable circulating clone. Monoclonal gammopathies that are associated with kidney disorders but lack an identifiable clone (ie, those that do not meet the criteria for myeloma or cancer) are categorized as monoclonal gammopathies of renal significance (MGRS). Some MPGNs are good examples of MGRS. [8]
In addition to circulating immune complexes becoming entrapped in the glomerular basement membrane (GBM), experimental evidence indicates that complexes may be formed in situ when antigens adhere to the GBM, and antibodies subsequently bind to these antigens. Formation of such immune complexes triggers the same cascade as described above.
Complement-mediated disease
Complement-dominant disease, or complement-mediated disease, is the classification of MPGN that has immunofluorescence and stains intensely and primarily for complement rather than immunoglobulin. This pattern is seen in both C3 glomerulonephritis (C3GN) and C3 dense deposit disease (C3 DDD), as well as in C4-dominant versions of the former.
C3 dense deposit disease
This is a systemic disorder, as evidenced by dense deposits in the kidney, splenic sinusoids, Bruch membrane of the retina, as well as its association with acquired partial lipodystrophy. [9, 10] It has a high rate of recurrence in allografts. However, dense deposit disease is associated with hypocomplementemia in only about half of cases.
The chemical composition and origin of the dense deposits are not known, although bright staining with thioflavine-T and wheat germ agglutinin suggests the presence of N-acetyl-glucosamine. It is also important to know that dense deposits can mask the presence of monoclonal immunoglobulins, which can be seen after digestion with pronase. One hypothesis is that the dense deposits themselves cause complement activation. [11] This hypothesis is supported by the tram-track distribution of C3 deposits along the basement membrane.
NFa is present in 80% of patients with dense deposit disease. NFa stabilizes the alternative pathway convertase and results in complement activation and chronic C3 consumption. Deficiency of factor H, functionally defective factor H, mutant factor H binding site of C3 (Marder disease), and presence of inhibitory or blocking factor H antibodies, described in MPGN, may lead to an accumulation of the alternative pathway convertase and chronic C3 consumption.
Partial lipoid dystrophy (PLD) is associated commonly with CDGN and the presence of NFa. Adipocytes produce adipsin, which is identical to complement factor D and is responsible for activating the preconvertase C3b,Bb. NFa causes a lysis of adipocytes that produce adipsin, and the distribution of fat atrophy in partial lipoid dystrophy follows variations in the amount of adipsin produced by adipocytes. By analogy, NFa may cause damage to glomerular cells that produce complement.
C3 glomerulonephritis
C3 glomerulonephritis is an entity with immunofluorescence findings of isolated glomerular C3 deposits. [12] C3 glomerulonephritis is similar in etiology to dense deposit disease, arising as a result of alternate complement activation or mutations in the complement-regulating proteins, but deposits tend to be in endocapilary and mesangial and of lesser intensity that in C3DDD. C3NeF is commonly present. C3 glomerulonephritis has been associated with antifactor H activity and monoclonal gammopathies.
Serum C3 levels are usually low, but they can be normal, along with normal C4 levels. [13] It is important to note that a normal serum C3 level does not exclude C3 glomerulonephritis. Dense deposit disease and C3GN both lack immunoglobulin staining on immunofluorescence and are the result of alternative pathway dysregulation. [14] In the literature, these 2 entities are often referred to as C3 glomerulopathy.
MPGN without immune complexes or complement staining
This category of membranoproliferative disease is defined by lack of an infectious or immunologic pathway or complement dysregulation and is therefore heterogeneous in nature. Common causes of MPGN pattern without complement or immunoglobulin staining include the following [3] :
-
Antiphospholipid antibody syndrome (APS)
-
Radiation
-
Transplant glomerulopathy
-
Sickle cell disease
-
Resolving/healing stage of atypical hemolytic-uremic syndrome/thrombotic thrombocytopenic purpura.
Etiology
The MPGN pattern of injury in the kidneys can be caused by various diseases. The immunofluorescence pattern can aid in identifiying the underlying disease.
Immunoglobulin/immune-complex mediated disease
Autoimmune diseases
Autoimmune diseases associated with a membranoproliferative pattern of kidney injury include the following:
-
Systemic lupus erythematosus (SLE)
-
Sjögren syndrome
-
Rheumatoid arthritis
-
Scleroderma
-
Celiac disease
-
Systemic sclerosis [15]
Chronic infections
The following chronic viral, bacterial, and protozoal infections are associated with a membranoproliferative pattern of kidney injury:
-
Bacterial – Endocarditis, infected ventriculoatrial (or jugular) shunt, multiple visceral abscesses, Mycoplasma, Mycobacterium, Staphylococcus
-
Protozoal – Malaria, schistosomiasis
Rare cases of MPGN have been reported with infections including Haemophilus parainfluenzae endocarditis, [18] anaplasmosis, [19] and nocardia. [20]
COVID-19 immunizations
Both relapse and de novo cases of MPGN have been reported following administration of Pfizer and AstraZeneca vaccines against COVID-19. [21, 22]
Paraprotein deposition diseases
Paraprotein deposition diseases that are associated with a membranoproliferative pattern of kidney injury include the following:
-
Glomerulonephropathies associated with cryoglobulinemia type I
-
Waldenström macroglobulinemia
-
Immunotactoid glomerulopathy
-
Immunoglobulin light-chain or heavy-chain deposition diseases
-
Fibrillary glomerulonephritis
-
Monoclonal gammopathy of unknown significance
Malignant neoplasms
Lymphoma, leukemia, and carcinoma are also associated with a membranoproliferative pattern of kidney injury.
Complement-mediated disease
Disorders of complement dysregulation can also cause membranoproliferative pattern of kidney injury. They can be classified as glomerulonephritis (GN) or dense deposit disease (DDD) —C3GN, C3-DDD, C4 -GN, C4-DDD—and may arise from the following:
-
Mutations in complement regulatory proteins
-
Mutations in complement factor
-
Antibodies to complement factors or regulatory proteins
Membranoproliferative pattern of injury with negative immunofluorescence
The following are chronic and recurrent thrombotic microangiopathies associated with a membranoproliferative pattern of kidney injury
-
Healing phase of hemolytic-uremic syndrome (HUS) and/or thrombotic thrombocytopenic purpura (TTP)
-
Syndromes of circulating antiphospholipid (anticardiolipin) antibodies
-
Radiation nephritis
-
Nephropathy associated with hematopoietic stem cell transplantation
-
Sickle cell anemia and polycythemia
-
Transplant glomerulopathy
-
Antitrypsin deficiency
Epidemiology
In the United States, membranoproliferative glomerulonephritis (MPGN) is observed in 6-12% of kidney biopsies performed to evaluate glomerular diseases. This entity accounts for 7% of children and 12% of adults with idiopathic nephrotic syndrome. Idiopathic/primary MPGN commonly affects children and young adults from 8 to 30 years of age. Secondary causes are more common in patients who present after 30 years of age.
Men and women are almost equally affected.
Prognosis
The main predictors of an adverse outcome in membranoproliferative glomerulonephritis (MPGN) are nephrotic syndrome and hypertension at presentation, low glomerular filtration rate (GFR) at 1 year, and older age. [23, 24] Histologic characteristics of crescent formation, interstitial fibrosis, tubular atrophy, and multiple sclerotic glomeruli indicate a poor prognosis. However, hypocomplementemia is not a predictor of disease severity or prognosis.
Kawasaki et al reported worse prognosis in pediatric patients with MPGN related to complement component C3 than in those with immune complex–mediated MPGN. In their study of 37 patients, those with C3-related MPGN were more likely to be nonresponsive to therapy or progress to end-stage kidney disease (ESKD). [25]
Similarly in a retrospective cohort study examining disease course in 111 patients with complement-mediated MPGN (C3G-87, DDD-24) the rate of progression to CKD, ESKD or death in 6 years was about 40% for both C3G and DDD. [26] The rate of progression was similar to that in other national registries.
In a study of all adult ESKD patients in Australia and New Zealand who commenced kidney replacement therapy from 1996 through 2016, rates of survival on dialysis and following kidney transplantation were comparable in the 456 patients with MPGN and the 12,660 patients with another form of glomerulonephritis. [27] However, the rate of post-transplant recurrence of MPGN remains high. Patients with recurrent MPGN in the allograft had the worst outcomes compared with any other glomerular disease, with 5-year graft survival of about 25-30%. [4, 28]

-
Glomerulus with lobular accentuation from increased mesangial cellularity. A segmental increase occurs in the mesangial matrix, and the peripheral capillary walls are thickened (hematoxylin and eosin stained section; original magnification × 250). Courtesy of John A. Minielly, MD.
-
Glomerulus with mesangial interposition producing a double contouring of basement membranes, which, in areas, appear to surround subendothelial deposits (Jones silver methenamine–stained section; original magnification × 400). Courtesy of John A. Minielly, MD.
-
Electron microscopy of prominent, glomerular, subendothelial, immune-type electron deposits (original magnification × 11,400). Courtesy of John A. Minielly, MD.
-
Electron microscopy of glomerular basement membrane, intramembranous, somewhat linear, electron dense deposit (ie, dense deposit disease; original magnification × 11,400). Courtesy of John A. Minielly, MD.
-
Immunofluorescent stained section. Intense, peripheral, glomerular, capillary loop deposition of immunoglobulin G (IgG) in an interrupted linear pattern corresponding to extensive subendothelial immune deposits (original magnification × 400). Courtesy of John A. Minielly, MD.
