Background
Lennox-Gastaut syndrome (LGS), also known as childhood epileptic encephalopathy, is a devastating pediatric epilepsy syndrome constituting 1%–4% of childhood epilepsies. [1] The syndrome is characterized by multiple seizure types, intellectual disability or regression, and abnormal findings on electroencephalogram (EEG), with paroxysms of fast activity and generalized slow spike-and-wave discharges (1.5–2 Hz) (see the image below).
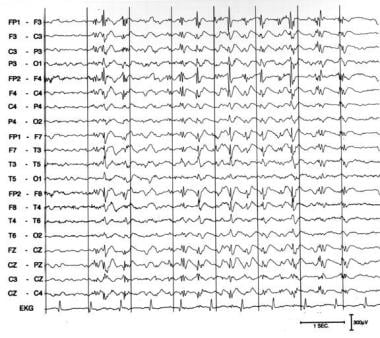 Slow spike wave pattern in a 24-year-old awake male with Lennox-Gastaut syndrome. The slow posterior background rhythm has frequent periods of 2- to 2.5-Hz discharges, maximal in the bifrontocentral areas, occurring in trains as long as 8 seconds without any clinical accompaniment.
Slow spike wave pattern in a 24-year-old awake male with Lennox-Gastaut syndrome. The slow posterior background rhythm has frequent periods of 2- to 2.5-Hz discharges, maximal in the bifrontocentral areas, occurring in trains as long as 8 seconds without any clinical accompaniment.
The most common seizure types are tonic-axial, atonic, and absence seizures, but myoclonic, generalized tonic-clonic, and partial seizures can be observed. An EEG is an essential part of the workup for LGS. Neuroimaging is an important part of the search for an underlying etiology.
A variety of therapeutic approaches are used in LGS, ranging from conventional antiepileptic agents to diet and surgery. Unfortunately, much of the evidence supporting these approaches is not robust, and treatment is often ineffective.
Etiology
Lennox-Gastaut syndrome (LGS) can be classified according to its suspected etiology as either idiopathic or symptomatic. Patients may be considered to have idiopathic LGS if normal psychomotor development occurred prior to the onset of symptoms, no underlying disorders or definite presumptive causes are present, and no neurologic or neuroradiologic abnormalities are found. In contrast, symptomatic LGS is diagnosed if a likely cause can be identified as being responsible for the syndrome.
Population-based studies have found that 70%–78% of patients with LGS have symptomatic LGS. Underlying pathologies in these cases may include the following:
-
Encephalitis and/or meningitis
-
Tuberous sclerosis
-
Brain malformations (eg, cortical dysplasias)
-
Hypoxia-ischemia injury
-
Frontal lobe lesions
-
Traumatic brain injury
There is a history of infantile spasms in 9%–39% of LGS patients.
In addition to idiopathic and symptomatic LGS, some investigators add “cryptogenic” as a third etiologic category. [3] This category encompasses cases in which the epilepsy appears to be symptomatic but a cause cannot be identified. In an epidemiologic study in Atlanta, Georgia, 44% of patients with LGS were in the cryptogenic group. In a series of 23 patients with cryptogenic LGS, 2.5%–47.8% had a family history of epilepsy and febrile seizures.
In 2001, the International League Against Epilepsy (ILAE) Task Force on Classification and Terminology proposed to include LGS among the epileptic encephalopathies. These are conditions in which not only the epileptic activity but also the epileptiform EEG abnormalities themselves are believed to contribute to the progressive disturbance in cerebral function. [4]
In the revision of the classification of seizures and epilepsy published in 2010, the terms "idiopathic," "symptomatic," and "cryptogenic" were eliminated and replaced by "genetic," "structural/metabolic," and "unknown" to encourage focus on specific underlying mechanisms. [5] However, many neurologists continue to use those terms as described above.
In 2022, the ILAE made three major changes to the definition of LGS compared with the traditional definition: (1) onset prior to 18 years, (2) must include tonic seizure, (3) generalized slow spike-waves (SSW) and (instead of or) generalized paroxysmal fast activity (GPFA) on electroencephalography (EEG). [6, 7]
Pathophysiology
The pathophysiology of Lennox-Gastaut syndrome (LGS) is not known. No animal models exist.
A variety of possible pathophysiologies have been proposed. One hypothesis states that excessive permeability in the excitatory interhemispheric pathways in the frontal areas is present when the anterior parts of the brain mature.
Involvement of immunogenetic mechanisms in triggering or maintaining some cases of LGS is hypothesized. Although one study [2] found a strong association between LGS and the human lymphocyte antigen class I antigen B7, a second study did not. No clear-cut or homogeneous metabolic pattern was noted in two separate reports of positron emission tomography (PET) studies in children with LGS.
Epidemiology
Overall, Lennox-Gastaut syndrome (LGS) accounts for 1%–4% of patients with childhood epilepsy but 10% of patients with onset of epilepsy when younger than 5 years. The prevalence of LGS in Atlanta, Georgia, was reported as 0.26 per 1000 live births. [1]
LGS is more common in boys than in girls. The prevalence is 0.1 per 1000 population for boys, versus 0.02 per 1000 population for girls (relative risk, 5.31).
The mean age at epilepsy onset is 26–28 months (range, 1 d to 14 y). The peak age at epilepsy onset is older in patients with LGS of an identifiable etiology than in those whose LGS has no identifiable etiology. The difference in age of onset between the group of patients with LGS and a history of West syndrome (infantile spasm) and those with LGS without West syndrome is not significant. The average age at diagnosis of LGS in Japan was 6 years (range, 2–15 y).
Epidemiologic studies in industrialized countries (eg, Israel, Spain, Estonia, Italy, Finland) have demonstrated that the proportion of epileptic patients with LGS seems relatively consistent across the populations studied and similar to that in the United States. The prevalence of LGS is 0.1–0.28 per 1000 population in Europe. The annual incidence of LGS in childhood is approximately 2 per 100,000 children. [8]
Among children with intellectual disability, 7% have LGS, while 16.3% of institutionalized patients with intellectual disability have LGS.
Prognosis
Long-term prognosis overall is unfavorable but variable in Lennox-Gastaut syndrome (LGS). [9] Longitudinal studies have found that a minority of patients with LGS eventually could work normally, but 47%–76% still had typical characteristics (intellectual disability, treatment-resistant seizures) many years after onset and required significant help (eg, home care, institutionalization). [10]
Patients with symptomatic LGS, particularly those with an early onset of seizures, prior history of West syndrome, higher frequency of seizures, or constant slow EEG background activity, have a worse prognosis than those with idiopathic seizures.
Tonic seizures may persist and be more difficult to control over time, while myoclonic and atypical absences appear easier to control.
The characteristic diffuse slow spike wave pattern of LGS gradually disappears with age and is replaced by focal epileptic discharges, especially multiple independent spikes.
Mortality rate is reported at 3% (mean follow-up period of 8.5 y) to 7% (mean follow-up period of 9.7 y). Death often is related to accidents. A high rate of injuries is associated with atonic and/or tonic seizures.
The severity of the seizures, frequent injuries, developmental delays, and behavior problems take a large toll on even the strongest parents and family structures. Pay attention to the psychosocial needs of the family (especially siblings). The proper educational setting also is important to help the patient with LGS reach his or her maximal potential.
Patient Education
Patients and their families need to be informed of the risk for the following severe idiosyncratic reactions from three commonly used antiepileptic medications for Lennox-Gastaut syndrome (LGS):
-
Valproate - Hepatotoxicity, pancreatitis
-
Lamotrigine -Stevens-Johnson syndrome, toxic epidermal necrolysis
-
Felbamate - Aplastic anemia, hepatotoxicity
-
Patient with Lennox-Gastaut syndrome wearing a helmet with face guard to protect against facial injury from atonic seizures
-
Slow spike wave pattern in a 24-year-old awake male with Lennox-Gastaut syndrome. The slow posterior background rhythm has frequent periods of 2- to 2.5-Hz discharges, maximal in the bifrontocentral areas, occurring in trains as long as 8 seconds without any clinical accompaniment.
