Practice Essentials
Limb-girdle muscular dystrophy (LGMD) refers to a group of genetic disorders that cause progressive weakness and wasting of the skeletal muscles, predominantly around the shoulders and hips.
Signs and symptoms
Most patients present with a history of progressive, symmetric, proximal muscle weakness that starts in childhood to young adulthood. Pelvic muscle weakness is most often the first symptom. Other features may include the following:
-
Asymmetric or distal weakness (rare)
-
Respiratory failure
-
Cardiomyopathy, cardiac arrhythmias
-
Scapular winging
-
Calf hypertrophy
-
Contractures
See Clinical Presentation for more detail.
Diagnosis
Muscle biopsy and genetic testing are the most important tools used in the diagnostic evaluation of patients in whom limb-girdle muscular dystrophy (LGMD) is suspected.
Serum creatine kinase level is complementary, and may be significantly elevated in some forms of LGMD, especially the autosomal recessive LGMDs.
Magnetic resonance imaging (MRI) of muscles can help differentiate some forms of LGMD.
See Workup for more detail.
Management
Although causative gene mutations have been well characterized for LGMD, no specific treatment is available for any of the LGMD syndromes yet.
Supportive care is essential to preserve muscle function, maximize functional ability, and prolong life expectancy.
Use of passive stretching, bracing, and orthopedic procedures allow the patient to remain independent for as long as possible.
Orthopedic surgery may be needed to help correct or prevent contractures and scoliosis.
See Treatment and Medication for more detail.
Background
Walton and Nattrass first proposed limb-girdle muscular dystrophy (LGMD) as a nosological entity in 1954. [1] Their definition included the following characteristics:
-
Expression in either male or female sex
-
Onset usually in the late first or second decade of life (but also middle age)
-
Usually autosomal recessive and less frequently autosomal dominant
-
Primary involvement of shoulder or pelvic-girdle muscles with variable rates of progression
-
Variable rates of progression
-
Severe disability within 20–30 years
-
Muscular pseudohypertrophy, contractures uncommon
Their definition was primarily reliant on phenotypic appearance, and thus included a heterogenous groups of disorders, including some that were not truly LGMD.
In 1995, an alphanumeric system of LGMD classification was introduced. This assigned a number based on mode of inheritance (1: autosomal dominant; 2: autosomal recessive), and an alphabet based on the order of discovery of linkage to a specific, certain genetic locus or a new disease gene. At the time of this writing, more than 30 genetic subtypes of LGMD have been identified. As the list continued to expand, a lack of consensus on nomenclature was evident, once classification exceeds LGMD 2Z. As of 2017, there are 34 types of LGMD detailed in the OMIM database.
Notably, LGMD subtypes are phenotypically highly variable, limb-girdle weakness may not be the predominant presentation, and mutation in genes assigned to LGMD subtypes may cause allelic conditions with a different phenotype. For example, mutations in TTN gene may present with a wide range of phenotypes ranging from congenital myopathy to late-onset distal myopathy. [2]
The 229th ENMC international workshop has proposed that for a condition to be considered LGMD, the following conditions must be fulfilled: [3]
-
Condition must be described in at least two unrelated families
-
Affected individuals must have achieved independent walking
-
Evidence of elevated serum creatine kinase (CK) activity
-
Presence of degenerative changes on muscle imaging over the course of the disease
-
Presence of dystrophic changes on muscle histology; development of end-stage pathology for the most affected muscles, over time
Application of this definition has led to exclusion of 10 conditions from the previous LGMD umbrella, including myofibrillar myopathy (LGMD1E). [3] The new proposed LGMD subtype classification system follows the formula: “LGMD, inheritance (R [recessive] or D [dominant]), order of discovery (number), affected protein.” In the absence of an identified pathogenic gene, phenotypic presentations that fulfill the above definition criteria are referred to as "LGMD unclassified."
Table 1. Conditions that are no longer considered LGMD, as per the definition proposed by the 229th ENMC international workshop, 2017 (Open Table in a new window)
| Previous name | Gene | Reason for exclusion | Proposed new name |
|---|---|---|---|
| LGMD1A | Myot | Distal weakness | Myofibrillar myopathy |
| LGMD1B | LMNA | High risk of cardiac arrhythmias; EDMD phenotype | Emery–Dreifuss muscular dystrophy (EDMD) |
| LGMD1C | CAV3 | Main clinical features rippling muscle disease and myalgia | Rippling muscle disease |
| LGMD1E | DES | Primarily false linkage; distal weakness and cardiomyopathy | Myofibrillar myopathy |
| LGMD1H | — | False linkage | Not confirmed |
| LGMD2R | DES | Distal weakness | Myofibrillar myopathy |
| LGMD2V | GAA | Histological changes | Pompe disease |
| LGMD2W | PINCH2 | Reported in a single family | PINCH2-related myopathy |
| LGMD2X | BVES | As above | BVES-related myopathy |
| LGMD2Y | TOR1AIP1 | As above | TOR1AIP1-related myopathy |
Table 2. New classification of LGMD with relevant affected protein (Open Table in a new window)
| LGMDD (previous name) | LGMDR (previous name) |
|---|---|
LGMD D1-DNAJB6; 7q36 [LGMD1E, 1D] LGMD D2-TNPO3; 7q32 [LGMD1F] LGMD D3-HNRPDL; 4q21 [LGMD1G] LGMD D4-Calpain-3; 15q15 [LGMD1I]
LGMD D5 (Bethlem): COL6A1: 21q22 COL6A2: 21q22 COL6A3: 2q37 |
R1 (2A): Calpain-3; 15q15 R2 (2B): Dysferlin; 2p13 R3 (2D): α-Sarcoglycan; 17q21 R4 (2E): β-Sarcoglycan; 4q12 R5 (2C): γ-Sarcoglycan; 13q12 R6 (2F): δ-Sarcoglycan; 5q33 R7 (2G): Telethonin; 17q12 R8 (2H): TRIM32; 9q33 R9 (2I; MDDGC5): FKRP; 19q13 R10 (2J): Titin; 2q24 R11 (2K; MDDGC1): POMT1; 9q34 R12 (2L): ANO5; 11p14 R13 (2M; MDDGC4): Fukutin; 9q31 R14 (2N; MDDGC2): POMT2; 14q24 R15 (2O; MDDGC3): POMGnT1; 1p32 R16 (2P; MDDGC9): DAG1; 3p21 R17 (2Q): Plectin 1f; 8q24 R18 (2S): TRAPPC11; 4q35 R19 (2T): GMPPB; 3p21 R20 (2U): ISPD; 7p21 R21 (2Z): POGLUT1; 3q13 R22: COL6A2; 21q22 R23: LAMA2; 6q22 R24: POMGNT2; 3p22 |
The old classification system, with reference to new nomenclaure, is retained here for description.
Although not truly limb-girdle syndromes, diseases classified as myofibrillar myopathies share several phenotypic characteristics with the LGMDs. They are usually adult-onset diseases with slowly progressive weakness involving proximal (and distal) muscles. Many patients have respiratory failure, cardiomyopathy, and neuropathy. Some mutations can cause both a myofibrillar myopathy and a muscular dystrophy phenotype. X-linked limb girdle dystrophies (dystrophinopathies, Emery–Dreifuss, McLeods Syndrome, and vacuolar) are described elsewhere.
Pathophysiology
LGMD is caused by mutations in genes encoding for proteins constituting the sarcolemma, cytosolic contents, or nucleus of muscle cells (myocytes). Given the heterogenous nature of mutations, mechanism of myocyte damage and muscle fiber degeneration may variably include errors in protein complex formation, functional or structural errors in the contractile apparatus, sarcolemmal instability, enzymatic abnormalities, or errors in repair mechanisms. With accumulating damage, there is eventual deposition and replacement of muscle by fibrotic and adipose tissue. Although the primary defect in many LGMDs is known, the precise mechanism leading to the dystrophic phenotype has not always been elucidated. Specific protein function and abnormalities are discussed below with each LGMD.
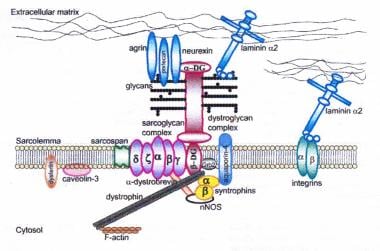 Dystrophin-glycoprotein complex bridges the inner cytoskeleton (F-actin) and the basal lamina. Mutations in all sarcoglycans, dysferlin, and caveolin-3, as well as mutations that cause abnormal glycosylation of alpha-dystroglycan can result in limb-girdle muscular dystrophy syndrome. Reprinted with permission from Cohn RD. Dystroglycan: important player in skeletal muscle and beyond. In: Neuromuscular Disorders. Vol. 15. Cohn RD. Elsevier; 2005: 207-17. 7, 20
Dystrophin-glycoprotein complex bridges the inner cytoskeleton (F-actin) and the basal lamina. Mutations in all sarcoglycans, dysferlin, and caveolin-3, as well as mutations that cause abnormal glycosylation of alpha-dystroglycan can result in limb-girdle muscular dystrophy syndrome. Reprinted with permission from Cohn RD. Dystroglycan: important player in skeletal muscle and beyond. In: Neuromuscular Disorders. Vol. 15. Cohn RD. Elsevier; 2005: 207-17. 7, 20
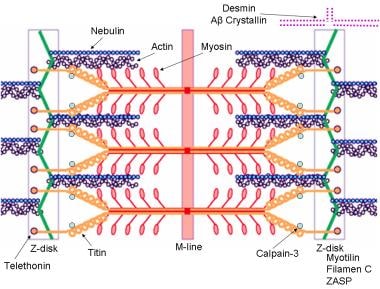 Schematic of the sarcomere with labeled molecular components that are known to cause limb-girdle muscular dystrophy or myofibrillar myopathy. Mutations in actin and nebulin cause the congenital myopathy nemaline rod myopathy, and the mutations in myosin cause familial hypertrophic cardiomyopathy. Image courtesy of Dr F. Schoeni-Affoher, University of Friberg, Switzerland.
Schematic of the sarcomere with labeled molecular components that are known to cause limb-girdle muscular dystrophy or myofibrillar myopathy. Mutations in actin and nebulin cause the congenital myopathy nemaline rod myopathy, and the mutations in myosin cause familial hypertrophic cardiomyopathy. Image courtesy of Dr F. Schoeni-Affoher, University of Friberg, Switzerland.
There is considerable overlap of the LGMD phenotype with other hereditary myopathies. Often, mutations in the same gene lead to phenotypes variably characterized as LGMD or as congenital muscular dystrophy, myofibrillar myopathy, or less commonly as Emery-Dreifuss muscular dystrophy, congenital myasthenic syndrome, congential myopathy, or metabolic myopathy. Most of these are discussed in separate chapters, but mutations causing myofibrillar myopathy are discussed in this article. Of interest, several mutations that result in myofibrillar myopathy are in genes that code for Z-disk proteins.
LGMD subtypes based on pathophysiological mechanism: [4]
-
Dystrophin-glycoprotein complex (Sarcoglycanopathy): LGMD2C-F
-
Sarcomeric proteins: LGMD1A, LGMD1D, LGMD1E, LGMD2A, LGMD2G, LGMD2J, LGMD2Q, LGMD2R
-
Defects in glycosylation/α-dystroglycan (α-dystroglycanopathy): LGMD2I, LGMD2K, LGMD2M, LGMD2N, LGMD2O, LGMD2P, LGMD2S, LGMD2T, LGMD2U, LGMD2Z
-
Proteins of nuclear envelope/function: LGMD1B, LGMD1F, LGMD1G, LGMD2X, LGMD2Y
-
Defects in signal transduction: LGMD1C, LGMD2P, LGMD2W
-
Defects in trafficking/repair: LGMD1C, LGMD1F, LGMD2B, LGMD2L
Frequency
Autosomal recessive LGMDs (LGMDR) are more common than the autosomal dominant forms of the disease (LGMDD), which probably account for about 10% of all LGMDs. The pooled prevalence of LGMD syndromes has been estimated to be 1.63 per 100,000 (range 0.56–5.75).
Different populations often have different frequencies of the various LGMDs.
Several studies throughout the world have estimated the frequency of LGMDs based on immunochemical and genetic testing. [5, 6, 7, 8, 9] In many studies, LGMD2A is the most common, accounting for 8–26% of all LGMDs. In some populations, it may be the only LGMD present (Reunion Island, Basque Country) with very high prevalence rates (48–69 cases per million). LGMD2B is also relatively common, accounting for 3–19% of all LGMDs. LGMD2I is common in certain parts of Northern Europe (Denmark and parts of England), but worldwide frequencies outside this area account for 3–8% of all LGMDs.
The sarcoglycanopathies as a group (LGMD2C-LGMD2F) are a common cause of LGMDs, accounting for 3–18%, with a high percentage of severe cases. As with other LGMDs, different sarcoglycanopathies are overrepresented or underrepresented in different populations, with some populations having representative cases of all 4 sarcoglycanopathies and other populations having only 1 mutation type, which is probably related to founder effects and population inbreeding (consanguinity). LGMD2C is common in Tunisia; LGMD2D is common in Europe, the United States, and Brazil; and LGMD2E and LGMD2F are common in Brazil. Overall, LGMD2D (α-sarcoglycanopathy) is twice as common as LGMD2C (γ-sarcoglycanopathy) and LGMD2E (β-sarcoglycanopathy), and LGMD2F (δ-sarcoglycanopathy) is the rarest.
All the congenital muscular dystrophies can present with a LGMD phenotype, and OMIM recognizes 4 at this time (LGMD2I, LGMD2K, LGMD2M, LGMD2N).
Mortality/Morbidity
Morbidity and mortality rates vary. In general, early onset forms tend to have a rapid course.
Patients with severe forms may become wheelchair bound in their early teens and die from respiratory complications in their late teens.
Patients with slowly progressive LGMD may retain independent ambulation into middle age. Some patients with confirmed mutations have had nearly normal strength.
Epidemiology
LGMD is reported in races and countries throughout the world. It is the fourth most common muscular dystrophy after dystrophinopathies, myotonic dystrophies, and fascioscapulohumeral dystrophy.
Autosomal dominant and autosomal recessive forms of LGMD affect both sexes equally.
The age of onset varies among the different mutations. It also can vary amongst families and family members with the same mutation. Reported age of onset of LGMDs is between 1 and 50 years, although some patients may be asymptomatic. Myofibrillar myopathies can present in the first decade of life up until the 60s or 70s.
Prognosis
The prognosis depends on the specific genetic mutation.
Pulmonary insufficiency, cardiomyopathy, and cardiac arrhythmia are the major causes of death.
Patient Education
Genetic counseling is often helpful to patients with limb-girdle muscular dytstrophy (LGMD) and their families to assist in family-planning decisions.
For additional information, see Muscular Dystrophy Association.
-
Dystrophin-glycoprotein complex bridges the inner cytoskeleton (F-actin) and the basal lamina. Mutations in all sarcoglycans, dysferlin, and caveolin-3, as well as mutations that cause abnormal glycosylation of alpha-dystroglycan can result in limb-girdle muscular dystrophy syndrome. Reprinted with permission from Cohn RD. Dystroglycan: important player in skeletal muscle and beyond. In: Neuromuscular Disorders. Vol. 15. Cohn RD. Elsevier; 2005: 207-17. 7, 20
-
Schematic of the sarcomere with labeled molecular components that are known to cause limb-girdle muscular dystrophy or myofibrillar myopathy. Mutations in actin and nebulin cause the congenital myopathy nemaline rod myopathy, and the mutations in myosin cause familial hypertrophic cardiomyopathy. Image courtesy of Dr F. Schoeni-Affoher, University of Friberg, Switzerland.
-
Top: Photomicrograph shows normal alpha-sarcoglycan staining of a myopathic biopsy specimen. Note dark staining around the rims of the muscle fibers. Bottom: Alpha-sarcoglycan stain of a muscle biopsy specimen from a patient with alpha-sarcoglycan deficiency. Note the absence of staining at the rims of the muscle fibers. Patterns of staining similar to these are observed in all the sarcoglycanopathies, dysferlinopathy, calpainopathy and limb-girdle muscular dystrophy type 2I (LGMD2I, Fukutin-related proteinopathy). However, staining may be variably reduced or absent.
-
Gomori trichrome–stained section in patient with myofibrillar myopathy. Note the abnormal accumulations of blue-red material in several muscle fibers.
-
Immunohistochemical staining by using an anti-desmin antibody in a patient with a myofibrillar myopathy. Courtesy of Alan Pestronk.
Tables
| Previous name | Gene | Reason for exclusion | Proposed new name |
|---|---|---|---|
| LGMD1A | Myot | Distal weakness | Myofibrillar myopathy |
| LGMD1B | LMNA | High risk of cardiac arrhythmias; EDMD phenotype | Emery–Dreifuss muscular dystrophy (EDMD) |
| LGMD1C | CAV3 | Main clinical features rippling muscle disease and myalgia | Rippling muscle disease |
| LGMD1E | DES | Primarily false linkage; distal weakness and cardiomyopathy | Myofibrillar myopathy |
| LGMD1H | — | False linkage | Not confirmed |
| LGMD2R | DES | Distal weakness | Myofibrillar myopathy |
| LGMD2V | GAA | Histological changes | Pompe disease |
| LGMD2W | PINCH2 | Reported in a single family | PINCH2-related myopathy |
| LGMD2X | BVES | As above | BVES-related myopathy |
| LGMD2Y | TOR1AIP1 | As above | TOR1AIP1-related myopathy |
| LGMDD (previous name) | LGMDR (previous name) |
|---|---|
LGMD D1-DNAJB6; 7q36 [LGMD1E, 1D] LGMD D2-TNPO3; 7q32 [LGMD1F] LGMD D3-HNRPDL; 4q21 [LGMD1G] LGMD D4-Calpain-3; 15q15 [LGMD1I]
LGMD D5 (Bethlem): COL6A1: 21q22 COL6A2: 21q22 COL6A3: 2q37 |
R1 (2A): Calpain-3; 15q15 R2 (2B): Dysferlin; 2p13 R3 (2D): α-Sarcoglycan; 17q21 R4 (2E): β-Sarcoglycan; 4q12 R5 (2C): γ-Sarcoglycan; 13q12 R6 (2F): δ-Sarcoglycan; 5q33 R7 (2G): Telethonin; 17q12 R8 (2H): TRIM32; 9q33 R9 (2I; MDDGC5): FKRP; 19q13 R10 (2J): Titin; 2q24 R11 (2K; MDDGC1): POMT1; 9q34 R12 (2L): ANO5; 11p14 R13 (2M; MDDGC4): Fukutin; 9q31 R14 (2N; MDDGC2): POMT2; 14q24 R15 (2O; MDDGC3): POMGnT1; 1p32 R16 (2P; MDDGC9): DAG1; 3p21 R17 (2Q): Plectin 1f; 8q24 R18 (2S): TRAPPC11; 4q35 R19 (2T): GMPPB; 3p21 R20 (2U): ISPD; 7p21 R21 (2Z): POGLUT1; 3q13 R22: COL6A2; 21q22 R23: LAMA2; 6q22 R24: POMGNT2; 3p22 |
