Background
Myasthenia gravis (MG) is a relatively rare autoimmune disorder in which antibodies form against nicotinic acetylcholine (ACh) postsynaptic receptors at the neuromuscular junction (NMJ) of the skeletal muscles, causing muscle weakness and rapid muscle fatigue. It is a type-II hypersensitivity immune response. The basic pathology is a reduction in the number of ACh receptors (AChRs) at the postsynaptic muscle membrane brought about by an acquired autoimmune reaction producing anti-AChR antibodies.
The reduction in the number of AChRs results in a characteristic pattern of progressively reduced muscle strength with repeated use and recovery of muscle strength after a period of rest. The ocular and bulbar muscles are affected most commonly and most severely, but most patients also develop some degree of fluctuating generalized weakness. [1] The most important aspect of MG in emergency situations is acute worsening of weakness leading to neuromuscular respiratory failure. The diagnosis of myasthenic versus cholinergic crisis and its management is also a significant challenge in emergent settings.
MG is a well-understood and well-managed disease. Pharmacologic therapy includes anticholinesterase agents, such as pyridostigmine, and immunosuppressive agents, such as corticosteroids, azathioprine, mycophenolate mofetil, tacrolimus, sirolimus, cyclosporine, cyclophosphamide, rituximab, plasmapheresis, and intravenous immune globulin (IVIg). Thymectomy has a significant role in the treatment of patients with generalized MG who are positive for acetylcholine receptor antibodies. Thymectomy becomes mandatory if a thymoma is present. Patients with MG require close follow-up care by a neurologist or neuromuscular specialist in cooperation with their primary care physician.
Anatomy
The neuromuscular junction (NMJ) serves as a transducer and amplifier to the peripheral nerve’s relatively small electrical current using a chemical signal subserved by neurotransmitter ACh. This in turn produces an electrical current of sufficient intensity and proper location such that it initiates a propagating action potential in the muscle fiber. Thus, the NMJ is considered a chemical synapse that functions as an electrical-chemical-electrical link.
In myasthenia gravis (MG), autoantibodies (immunoglobulin G [IgG1]) develop against nicotinic acetylcholine postsynaptic receptors at the NMJ of skeletal muscles. [2, 3] The reasons for this development are unknown, although it is clear that certain genotypes are more susceptible. [4] To understand MG, it is necessary to become familiar with the normal anatomy and physiology of the NMJ (see the image below). A full appreciation of the normal function of the NMJ is needed to understand the principles underlying diagnostic testing and the mechanisms of therapeutic interventions.
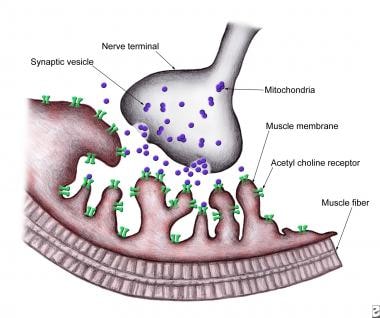 Normal neuromuscular junction showing a presynaptic terminal with a motor nerve ending in an enlargement (bouton terminale): Synaptic cleft and postsynaptic membrane with multiple folds and embedded with several acetylcholine receptors.
Normal neuromuscular junction showing a presynaptic terminal with a motor nerve ending in an enlargement (bouton terminale): Synaptic cleft and postsynaptic membrane with multiple folds and embedded with several acetylcholine receptors.
The nerve terminal of the motor nerve enlarges at its end to form the so-called terminal bouton or terminal bulb. This bulbous nerve terminal lies within a groove or indentation along the somatic muscle fiber. The NMJ can be subdivided into 3 major areas: presynaptic region, synpatic space, and post-synaptic membrane. Disorders of neuromuscular transmission can arise from abnormalities in each of these locations.
The pre-synaptic region
Each motor neuron has an axon that branches distally to provide nerve terminals that innervate muscle fibers through the NMJ. Collectively, they are known as the motor unit. A muscle fiber is innervated by only one motor neuron with the exception of extraocular muscle fibers where single muscle fibers may receive multiple innervation. The motor nerve loses its myelin sheath as it approaches near the NMJ where it begins to divide into terminal branches. Each terminal branch of an axon, as it nears an individual muscle fiber, expands into a presynaptic terminal bouton that lies in a depression in the muscle membrane. A basement membrane overlies this terminal interface between the bouton and its muscle fiber, constituting part of the muscle end-plate. The terminal bouton has a number of subcellular components including neurotubules, neurofilaments, multiple mitochondria, and a large number of membrane-bound vesicles ranging from 300 to 500 Angstrom units in diameter, called synaptic vesicles. Each vesicle contains approximately 1 quantum of acetylcholine, which equals 10,000 molecules of acetylcholine. A single nerve terminal has approximately 200,000 synaptic vesicles. These vesicles are organized in 3 discrete groups.
The primary, or immediately available store consists of approximately 1000 quanta of ACh and are located just beneath the pre-synaptic nerve terminal membrane at specific sites called active zones. These release sites (active zones) lie directly opposite the ACh receptors (AChRs) located on the post-synaptic membrane. The primary store is available for immediate release. They behave like soldiers at the front line, ready for action.
The secondary, or mobilization store consists of approximately 10,000 quanta of ACh that can replenish the primary store after a few seconds. These behave like immediate reinforcements that become mobilized to replace the depleted primary store.
Finally, a tertiary, or reserve store of more than 100,000 quanta exists far from the NMJ in the axon and cell body. They behave like the reserves.
Voltage-gated calcium (Ca++) channels (P/Q-type) are located in the proximity of the active zones. Under the electron microscope they appear as double parallel rows of dense intramembrane particles.
The synaptic space
Located between the presynaptic region and the post-synaptic muscle membrane lies the synaptic left, which is 50–75 nm wide. It comprises the primary cleft and a number of secondary clefts (subneural clefts). The synaptic cleft is bound laterally by a basement membrane. The primary synaptic cleft has AChR concentrated at the crests of postsynaptic folds opposite the presynaptic active zones. Secondary synaptic clefts (subnerual clefts) are also a significant part of the post-synpatic membrane and located in its depth are present voltage-gated sodium channels.
The postsynaptic membrane
The postsynaptic membrane is a highly specialized area of the muscle fiber membrane, known as the end-plate. The end-plate is a convoluted structure with numerous junction folds enhancing the surface area of the post-synaptic membrane. On the crests of each junctional fold are AChR clusters aligned in proximity with an active zone on the presynaptic terminal. The basal lamina of the endplate contains the enzyme acetylcholinesterase (AChE), which are located in the troughs of the junctional folds. [5] The concentration of AChE is five- to eight-fold lower than concentration of ACh receptors, but is enough to hydrolyze most of the ACh release by the nerve terminal and prevents repeated binding of ACh to AChRs. Voltage-gated sodium channels are present in large numbers on the post-synaptic membrane and are particularly concentrated in the depths of the secondary synaptic clefts.
The acetylcholine receptor
AChR is an ionotropic ligand-gated transmembrane receptor channel. It is a quaternary glycoprotein structure with 5 subunits surrounding a central cation channel with negatively charged inner wall. In the adult or "innervated" form of AChR (2α1ß1δ1ε) and in the fetal or "denervated" form of AChR (2α1ß1δ1γ). It has a half-life of 8–11 days. [6] These subunits are homologous across different species, suggesting that the encoding genes evolved from a common ancestral gene. The AChR subunits are arranged in a circle, spanning the membrane forming a central opening that acts as an ion channel (see the image below). Each AChR subunit is composed of four transmembrane domains (M1–M4).
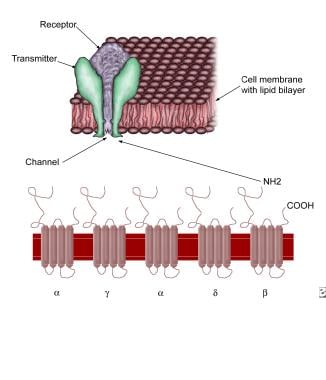 Acetylcholine receptor. Note 5 subunits, each with 4 membrane-spanning domains forming a rosette with a central opening. The central opening acts as an ion channel.
Acetylcholine receptor. Note 5 subunits, each with 4 membrane-spanning domains forming a rosette with a central opening. The central opening acts as an ion channel.
The subunits in the AChRs are organized like barrel staves with their convexity inward or in a funnel-like fashion with the narrow end oriented to the intracellular compartment. In the center of the funnel-like portion of the AChR is a cation channel, which is contributed by the M2 and M3 domains of each subunit. Negatively charged residues located at either end of the channel allow the passage of positive ions and exclude passage of the negative ions. Ion binding sites within the channel have an important role in the passage of ions. The ACh binding sites and the main immunogenic region (MIR) are located on the protruding extracellular surfaces of alpha subunits; although in close proximity, the sites for MIR and ACh are distinctly separate. The AChR binding sites are actually located between the α1 and δ subunits and between the α2 and ε subunits. The extracellular portions of AChR extend 100 Angstrom units beyond the cell membrane. Sugars on the extracellular surface of the subunits extend outward and have complex branching patterns. On the cytoplasmic surface of the membrane are cytoskeletal components, which anchor the AChRs. The post-synaptic junctional folds are packed with AChRs (10,000 receptors/µm2) and also contain other protein subsets like MuSK, LRP4, rapsyn, integrins, ErbB receptors, N-acetylgalactosaminlyl transferase, and collagen XIII. [7, 8] Complex interactions between agrin, rapsyn, and MuSK, and LRP4 are involved in the development and maintenance of the NMJ. The clustering of AChR at the crests of the postsynaptic junctional fold is critical for normal neuromuscular transmission.
The troughs of the junctional folds have neural cell adhesion molecules (NCAM), and the voltage-gated sodium ion channels. The latter are tethered to Ankyrin G and β-spectrin, and linked to the cytoskeleton by syntrophins. [9, 10] At the endplate, rapsyn connects AChRs to each other and muscle fiber cytoskeleton via dystrophin-glycoprotein complex (DGC). The DGC contains a number of transmembrane proteins (α- and β-dystroglycan and the sarcoglycan complex). DGC also has submembrane proteins (dystrophin, utrophin, syntrophin, and dystrobrevin) and connects to the cytoskelton via F-actin and to the basal lamina via laminin.
Neureregulin produced by the NR-1 gene is a motor nerve-derived trophic factor similar to agrin. It is thought to induce accumulation of voltage-gated sodium ion channels in the depths of the synaptic clefts. It is noted that shortly after neuregulin appears at the developing NMJ, fetal AChRs containing the γ-subunit (γ-AChRs) are replaced by adult AChRs containing the ε subunit (ε-AChRs). The ε subunit is dependent on the presence of neuregulin for its continued expression at the NMJ. [7] The mature AChRs are constantly turned over by internalization and degradation and replacement by new AChRs. They are not recycled.
Pathophysiology
The following physiologic events occur in neuromuscular transmission:
A nerve action potential propagates down the axon and depolarizes the presynaptic nerve terminal.
Voltage-gage calcium channels open in response to depolarization and there is calcium influx through these channels into the nerve terminal.
Synaptic vesicles fuse with the presynaptic membrane, releasing ACh into the synaptic space.
ACh molecules bind to AChR on the post-synaptic membrane causing the the recepors to undergo conformational change, opening the ion channel.
Membrane conductance to Na+ ions increases (Na+ goes in, K+ goes out) resulting in depolarization of endplate region causing end-plate potenital (EPP).
If the EPP is sufficient to depolarize the adjacent muscle membrane to threshold, an action potential is generated in the muscle fiber. Conversely, if it is of insufficient magnitude it will not cause the muscle membrane to reach threshold, and therefore fails to generate a muscle fiber action potential.
In the resting state, there is an intermittent release of ACh molecules across the primary synaptic cleft. The ACh molecules serve as ligands and bind to the AChR on the post-synaptic membrane. Two molecules of ACh bind to the alpha subunits of the AChR, and fuse with it. The bound AChR then undergoes a 3-dimensional conformational change in the central ion channel portion of the AChR (M2, alpha-helical) opening the funnel-shaped cation channel (whose inner wall is negatively charged). This opening is very brief (about 1 ms) resulting in influx of sodium ions while simultaneously allowing potassium efflux along its opposite concentration gradient. This results in brief depolarization of the muscle membrane only at the junctional region creating a post-synaptic non-propagating depolarization called miniature endplate potentials (MEPPs).
The normal muscle fiber resting membrane potential is -80 mV (negative inside). The threshold for triggering an action potential in the muscle fiber is -50mV to -65mV. When a nerve action potential depolarizes the terminal axons, sodium ion conductance is increased and at the same time voltage-gated calcium channels (VGCCs) are activated, allowing an influx of calcium ion at the terminal portion of the axon. The entry of calcium ions is critical to the process of neuromuscular transmission (if Ca++ is removed from the extracellular space, NMJ transmission ceases). The entry of Ca++ starts a complicated interaction of many proteins including SNARE protein complex at the nerve terminal leading to facilitation of fusion of the ACh-containing vesicles with the presynaptic membrane. Consequently, the discharge of ACh occurs by exocytosis into the synaptic cleft. The greater the calcium concentration inside the presynaptic terminal, the more quanta of ACh are released into the synaptic cleft.
ACh molecules bind to the AChRs resulting in a larger depolarization of the post-synaptic membrane resulting in the endplate potential (EPP). The amplitude of EPP is normally high enough to trigger an action potential at the post-synaptic membrane. The voltage-gated sodium channels present in the depths of the secondary synaptic cleft facilitate action potential, which is propagated along the NMJ muscle membrane. When this action potential invades the transverse tubule system of the muscle, another voltage-gated calcium channel (VGCC) becomes activated causing influx of calcium ions, and triggering mechanical contraction of the muscle fiber contractile apparatus.
The action of ACh on the post-synaptic membrane is short-lived and is terminated within a few milliseconds of its release from the nerve terminal through hydrolysis by the enzyme acetylcholinesterase into acetic acid and choline. The latter is taken up by the presynaptic membrane and repackaged into new ACh molecules.
The calcium ions are normally pumped out of the terminal portion of the axon within 100 ms, so they linger for a while and maintain the axon terminal in a hyperexcitable state, enhancing the release of ACh should a second action potential depolarizes the axon within this time frame.
Safety factor
The amplitude of the EPP tends to be >60 mV above the muscle fiber resting membrane potential of -80 mV. So only 15mV is needed to reach the threshold for action potential of -65 mV. The extra 45 mV is referred as the Safety Factor. So, even if the EPP were to get smaller (e.g., 40 mV) due to repetitive contraction resulting in fatigue, the EPP would still be high to reach threshold and maintain the one-to-one relationship between the action potential of the motor axon and generation of an action potential in the muscle cell. However, if the safety factor is greatly decreased as it may occur in MG, neuromuscular transmission may become blocked.
In myasthenia gravis (MG), the safety factor is reduced (i.e., baseline EPP is reduced but still above threshold). Slow RNS (3 Hz) will cause depletion of ACh quanta and may drop the EPP below threshold, resulting in the absence of a muscle fiber action potential (a phenomenon referred to as presynaptic rundown). Consequently, EPP is reduced as there are fewer AChRs for ACh molecules to bind.
In MG, there are anti-AChR-ab against AChR available at the post-synaptic folds, which become flattened or simplified by the immunopathological mechanism. This, on top of the gradual reduction of AChRs that are released with repeated use of the muscle, leads to insufficient endplate potentials (EPP), which may fall below the threshold value for generation of an action potential. The end result of this process is inefficient neuromuscular transmission. When this failure occurs at enough muscle fibers, it can manifest clinically and can be demonstrated electrophysiologically through low-frequency repetitive nerve stimulation (3 Hz RNS). However, if it occurs in only a few muscle fibers it can be detected on single-fiber electromyography (SFEMG).
Patients become symptomatic once the number of AChRs is reduced to approximately 30% of normal. The cholinergic receptors of smooth and cardiac muscle have a different antigenicity than skeletal muscle and usually are not affected by the disease.
The decrease in the number of postsynaptic AChRs is believed to be due to an autoimmune process whereby anti-AChR antibodies are produced and block the AChR. It causes an increase in the turnover of the AChR, and damage of the postsynaptic membrane in a complement-mediated manner.
The exact mechanism of loss of immunologic tolerance to AChR, a self-antigen, is not understood. MG can be considered a B cell–mediated disease, in that it derives from antibodies (a B cell product) against AChR. However, the importance of T cells in the pathogenesis of MG is becoming increasingly apparent. The thymus is the central organ in T cell–mediated immunity, and thymic abnormalities such as thymic hyperplasia or thymoma are well recognized in myasthenic patients.
It is thought that both the initiation and maintenance of MG occurs in a process that involves type-II hypersensitivity reactions. The production of autoantibodies implies that it is a B-cell-mediated autoimmune disorder. The intiation of the process is dependent on T-cell help. Accordingly, CD4 T cells are the main driving force in the immunopathogenesis of MG. Macrophages and dendritic cells are activated and these act as antigen-presenting cells. AChRs phagocytized by macrophages become degraded into peptide subcomponents. These are then linked to MHC-II, the molecule required for reactivity to “self-antigens.” The AChR antigenic fragment and MHC complex are transported to the surface of macrophages and dendritic cells. Specific, helper T cells, with the cooperation of CD3 complex and CD4 molecular T-cell receptor site, recognize this antigen complex. Specific receptor sites on the T-cell surface recognize cytokines secreted by the macrophage and dendritic cells. The activated helper T cells secrete interferon (IFN)-γ and interleukin (IL)-17 that stimulate B lymphocytes. The activated B lymphocytes grow and undergo clonal expansion into antibody-synthesizing plasma cells. These plasma cells secrete IgG anti-AChR antibodies that bind to the nictonic ACh-R.
Antibody response in MG is polyclonal. In an individual patient, antibodies are composed of different subclasses of IgG. In most instances, they are of IgG1 and IgG3 subclass and are directed against the main immunogenic region (MIR) on the alpha subunit. The alpha subunit is also the site of ACh binding, though the binding site for ACh is not always the same as the MIR. Binding of AChR antibodies to AChR results in impairment of neuromuscular transmission in several ways, including the following:
-
Cross-linking 2 adjacent AChRs with anti-AChR antibody, thus accelerating internalization and degradation of AChR molecules
-
Causing complement-mediated destruction of junctional folds of the postsynaptic membrane
-
Blocking the binding of ACh to AChR
-
Decreasing the number of AChRs at the NMJ by simplification of the junctional folds on the postsynaptic membrane, thereby reducing the surface area available for insertion of newly synthesized AChRs
Patients without anti-AChR antibodies are recognized as having seronegative MG (SNMG). These patients usually have autoantibodies (IgG4) against muscle-specific kinase (MuSK). These do not activate complement, unlike AChR-abs. MuSK plays a critical role in postsynaptic differentiation and clustering of AChRs at the NMJ to promote efficient neuromuscular transmission. MuSK autoantibodies are pathogenic and this is proven by passive transfer and active immunization studies in animals. It is likely MuSK autoantibodies disrupt the interaction between MuSK and the LRP4 and collagen Q. Patients with anti-MuSK antibodies are predominantly women, with a tendency of disease onset in the third or fourth decades. They have prominent oculobulbar weakness with dysarthria. Face and tongue atrophies have been reported in long-standing disease. It is often confused with bulbar amyotrophic lateral sclerosis (ALS). Another group has reported patients who exhibit prominent neck (dropped head syndrome), shoulder, and respiratory weakness. [11, 12] Myasthenic crises appear to be more frequent in patients with MuSK antibodies. [13, 14]
The role of the thymus in the pathogenesis of MG is not entirely clear, but 75% of patients with MG have some degree of thymus abnormality (eg, hyperplasia or thymoma). Histopathologic studies have shown prominent germinal centers. Epithelial myoid cells normally present in the thymus resemble skeletal muscle cells and possess AChRs on their surface membrane. These cells may become antigenic by molecular mimicry and unleash an autoimmune attack on the muscular endplate nicotinic AChRs.
The question of why MG affects the extraocular muscles first and predominantly remains unanswered. The answer probably has to do with type and distribution of NMJs in at least some of the muscles affected by the disease.
The distinctive features in MG, particularly the fluctuating nature of a patient's strength, is attributed to the unique pathophysiology of impaired neuromuscular transmission. This pathophysiology produces a dynamic rather than a fixed disorder as a result of the relative ease by which NMJs repair.
Etiology
Myasthenia gravis (MG) is idiopathic in most patients. Although the main cause behind its development remains speculative, the end result is a derangement of immune system regulation. MG is clearly an autoimmune disease in which the specific antibody has been characterized completely. In as many as 90% of generalized cases, IgG to AChR is present. [15] Even in patients who do not develop clinical myasthenia, anti-AChR antibodies can sometimes be demonstrated.
Patients who are negative for anti-AChR antibodies may be seropositive for antibodies against MuSK. Muscle biopsies of these patients show myopathic features with prominent mitochondrial abnormalities, as opposed to the neurogenic features and atrophy frequently found in MG patients positive for anti-AChR. The mitochondrial impairment could explain the oculobulbar involvement in anti-MuSK–positive MG. [16]
Numerous findings have been associated with MG. For example, people with certain human leukocyte antigen (HLA) types have a genetic predisposition to autoimmune diseases. The histocompatibility complex profile includes HLA-A1, -A3, -B7, -B8, -DRw3, and -DQw2 (though these have not been shown to be associated with the strictly ocular form of MG). However, HLA genotyping is not routinely used in the evaluation of patients suspected to have MG.
Sensitization to a foreign antigen that has cross-reactivity with the nicotinic ACh receptor has been proposed as a cause of myasthenia gravis, but the triggering antigen has not yet been identified.
Various drugs may induce or exacerbate symptoms of MG, including the following:
-
Antibiotics (eg, aminoglycosides, polymyxins, ciprofloxacin, erythromycin, and ampicillin)
-
Penicillamine - This can induce true myasthenia, with elevated anti-AChR antibody titers seen in 90% of cases; however, the weakness is mild, and full recovery is achieved weeks to months after discontinuance of the drug
-
Beta-adrenergic receptor blocking agents (eg, propranolol and oxprenolol)
-
Lithium
-
Magnesium
-
Procainamide
-
Verapamil
-
Quinidine
-
Chloroquine
-
Prednisone
-
Timolol (ie, a topical beta-blocking agent used for glaucoma)
-
Anticholinergics (eg, trihexyphenidyl)
-
Neuromuscular blocking agents (eg, vecuronium and curare) - These should be used cautiously in myasthenic patients to avoid prolonged neuromuscular blockade
-
Nitrofurantoin has also been linked to the development of ocular MG in 1 case report; discontinuance of the drug resulted in complete recovery.
-
MG induction as a side effect of cancer immunotherapy
Ipilimumab induced (anti-CTLA4) MG
PD-1, PDL-1 inhibitors
Thymic abnormalities are common: Of patients with MG, 75% have thymic disease, 85% have thymic hyperplasia, and 10–15% have thymoma. Extrathymic tumors may include small cell lung cancer and Hodgkin disease. [17, 18] Hyperthyroidism is present in 3–8% of patients with MG and has a particular association with ocular MG.
Epidemiology
In the United States in 2021, the overall incidence of myasthenia gravis (MG) was 3.2 per 100,000 with similar estimates for males and females (3.2 vs. 3.1 per 100,000, respectively). [19] Total prevalence was estimated to be 37.0 per 100,000 with sex-specific estimates being comparable at 37.3 and 36.7 per 100,000 for males and females, respectively. [19] These figures suggest that cases of MG are increasing, likely due to increased awareness and diagnosis of the disease.
Approximately 15%–20% of patients with MG experience crisis in their lifetime, typically within the first 2 years of the diagnosis. [18]
Fifty years ago, estimates of mortality in MG crisis ranged from 50% to 80%. [20, 21, 22] Currently, the overall in-hospital mortality rate is 2.2%, being higher in MG crisis (4.47%).
Age-related demographics
MG can occur at any age. Female incidence peaks in the third decade of life, whereas male incidence peaks in the sixth or seventh decade. The mean age of onset is 28 years in females and 42 years in males.
Transient neonatal MG occurs in infants of myasthenic mothers who acquire anti-AChR antibodies via placental transfer of IgG. Some of these infants may suffer from transient neonatal myasthenia due to effects of these antibodies.
Most infants born to myasthenic mothers possess anti-AChR antibodies at birth, yet only 10-20% develop neonatal MG. This may be due to protective effects of alpha-fetoprotein, which inhibits binding of anti-AChR antibody to AChR. High maternal serum levels of AChR antibody may increase the chance of neonatal MG; thus, lowering the maternal serum titer during the antenatal period by means of plasmapheresis may be useful.
Sex-related demographics
Classically, the overall female-to-male ratio has been considered to be 3:2, with a female predominance in younger adults (ie, patients aged 20-30 years) and a slight male predominance in older adults (ie, patients older than 50 years). [23, 1] Studies show, however, that with increased life expectancy, males are coming to be affected at the same rate as females. Ocular MG shows a male preponderance. The male-to-female ratio in children with MG and another autoimmune condition is 1:5. MuSK myasthenia is found predominantly in women and has a peak incidence of less than 40 years of age. [24]
Race-related demographics
The onset of MG at a young age is slightly more common in Asians than in other races. [23]
The annual incidence rate of MG is higher in black women (0.01 per 1000 persons/year) compared to white women (0.009 per 1000 persons/year) and white (0.008 per 1000 persons/year) and black men (0.007 per 1000 persons/year). [25]
MG can be classified by age of onset, site or sites of involvement on the NMJ, serological status, and associated thymic pathology. This is helpful in diagnosis and management. Also, the sensitivity of diagnostic tests may very according to age of onset, specific antibodies, and whether the disease is ocular or generalized.
The vast majority of patients have MG that is immune mediated, but they are very rare congenital myasathenic syndromes that have no immune basis and where immunosuppresants must not be used.
Prognosis
Given current treatment, which combines cholinesterase inhibitors, immunosuppressive drugs, plasmapheresis, immunotherapy, and supportive care in an intensive care unit (ICU) setting (when appropriate), most patients with myasthenia gravis (MG) have a near-normal life span. Mortality is now 3–4%, with principal risk factors being age older than 40 years, short history of progressive disease, and thymoma; previously, it was as high as 30–40%. In most cases, the term gravis is now a misnomer.
Morbidity results from intermittent impairment of muscle strength, which may cause aspiration, increased incidence of pneumonia, falls, and even respiratory failure if not treated. [15] In addition, the medications used to control the disease may produce adverse effects.
Today, the only feared condition arises when the weakness involves the respiratory muscles. Weakness might become so severe as to require ventilatory assistance. Those patients are said to be in myasthenic crisis.
The disease frequently presents (40%) with only ocular symptoms. However, the extraocular almost always are involved within the first year. Of patients who show only ocular involvement at the onset of MG, only 16% still have exclusively ocular disease at the end of 2 years.
In patients with generalized weakness, the nadir of maximal weakness usually is reached within the first 3 years of the disease. As a result, half of the disease-related mortality also occurs during this period. Those who survive the first 3 years of disease usually achieve a steady state or improve. Worsening of disease is uncommon after 3 years.
Thymectomy results in complete remission of the disease in a number of patients. However, the prognosis is highly variable, ranging from remission to death.
Patient Education
Educate patients with myasthenia gravis (MG) to recognize and immediately report impending respiratory crisis. Intercurrent infection may worsen symptoms of MG temporarily. Mild exacerbation of weakness is possible in hot weather.
The risk of congenital deformity (arthrogryposis multiplex) is increased in offspring of women with severe MG. Neonates born to women with MG must be monitored for respiratory failure for 1-2 weeks after birth. Certain immunosuppressant drugs have teratogenic potential. Discuss these aspects with women in reproductive years before beginning therapy with these drugs.
Certain medications (eg, aminoglycosides, ciprofloxacin, chloroquine, procaine, lithium, phenytoin, beta-blockers, procainamide, and quinidine) may exacerbate symptoms of MG; many others have been associated only rarely with exacerbation of MG. Patients should always consult a neurologist before starting any of these medications.
-
Normal neuromuscular junction showing a presynaptic terminal with a motor nerve ending in an enlargement (bouton terminale): Synaptic cleft and postsynaptic membrane with multiple folds and embedded with several acetylcholine receptors.
-
Acetylcholine receptor. Note 5 subunits, each with 4 membrane-spanning domains forming a rosette with a central opening. The central opening acts as an ion channel.
-
CT scan of chest showing an anterior mediastinal mass (thymoma) in a patient with myasthenia gravis.
-
Increasing left ptosis developing upon sustained upward gaze in patient with myasthenia gravis (A through F). Note limited elevation of left eye, denoting superior rectus palsy (A). A initially, C after around 20 seconds, F after 1 minute.
-
Cogan sign. Patient changes gaze from downward position (A) to primary position (B). Both lids are seen to overshoot in twitch (B) before gaining their initial ptotic position (D). In this case, Cogan sign is seen more obviously on right, whereas left lid is more ptotic.
-
CT scan of chest and mediastinum showing thymoma in patient with myasthenia gravis.
-
Repetitive nerve stimulation at frequency of 2 Hz showing increasing decrement in amplitude of compound muscle action potential up to fourth response (42% amplitude loss), after which it stabilizes.
-
Single-fiber electromyography showing so-called jitter phenomenon (second action potential wave group).
-
What is myasthenia gravis? Myasthenia gravis is an autoimmune disease that's categorized as a type II hypersensitivity that involves autoantibodies binding acetylcholine receptors on skeletal muscle cells. Courtesy of Osmosis.org (https://www.osmosis.org/).
-
Motor end plate and innervation. Courtesy of Wikimedia Commons.
Tables
| Distribution of Weakness | Percentage of Patients |
|---|---|
| Localized ocular | 17% |
| Ocular and generalized | 50% |
| Ocular and bulbar | 13% |
| Ocular and limb | 20% |







