Background
Systemic sclerosis (SSc) is a systemic connective tissue disease. Characteristics of systemic sclerosis include essential vasomotor disturbances; fibrosis; subsequent atrophy of the skin (see the image below), subcutaneous tissue, muscles, and internal organs (eg, alimentary tract, lungs, heart, kidney, CNS); and immunologic disturbances accompany these findings.
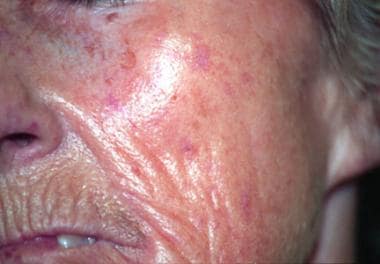 Telangiectasias affecting the face: They are pronounced and numerous, especially in the atrophic phase of the disease. Radical furrowing around the mouth is also characteristic in the later stage of the disease.
Telangiectasias affecting the face: They are pronounced and numerous, especially in the atrophic phase of the disease. Radical furrowing around the mouth is also characteristic in the later stage of the disease.
In one survey one quarter of patients with Raynaud phenomenon not fitting the 1980 American College of Rheumatology classification criteria were reclassified as having systemic sclerosis using the 2013 American College of Rheumatology/European League Against Rheumatism (EULAR) criteria. [1]
See Cutaneous Clues to Accurately Diagnosing Rheumatologic Disease, a Critical Images slideshow, to help recognize cutaneous manifestations of rheumatologic diseases.
Also see Juvenile Systemic Sclerosis for a pediatric focus.
Pathophysiology
Excessive collagen deposition causes skin and internal organ changes. Many factors, including environmental factors, can lead to immunologic system disturbances and vascular changes. Endothelial alterations may lead to a cascade of stimulatory changes that involve many cells, including fibroblasts, T lymphocytes, macrophages, and mast cells. In turn, the activated cells secrete a variety of substances, including cytokines and their soluble receptors and enzymes and their inhibitors. These substances lead to changes in the extracellular matrix compounds, including fibronectin; proteoglycans; and collagen types I, III, V, and VII. Increased collagen deposition in tissues is a characteristic feature of systemic sclerosis. Increased collagen production or disturbances in its degradation can cause excessive collagen deposition in tissues.
Fibrosis can be caused by profibrotic cytokines, including transforming growth factor-beta (TGF-beta), interleukin-4 (IL-4), platelet-derived growth factor (PDGF), and connective-tissue growth factor. [2] The vasculopathy may be linked to TGF-beta and PDGF, while the diminution of lesional cutaneous blood vessels can be attributed to antiendothelial cell autoantibodies. The activation of the immune system is of paramount importance in the pathogenesis of systemic sclerosis. Antigen-activated T cells, activated infiltrate early, infiltrate the skin, and produce the profibrotic cytokine IL-4. B cells may contribute to fibrosis, as deficiency of CD19, a B-cell transduction molecule, results in decreased fibrosis in animal models.
Different factors, including genetic, environmental, vascular, autoimmunologic, and microchimeric factors are involved in systemic sclerosis pathogenesis. One theory states that antigens from the human leukocyte antigen (HLA) histocompatability complex, including HLA-B8, HLA-DR5, HLA-DR3, HLA-DR52, and HLA-DQB2, are involved in systemic sclerosis. Some data suggest that apoptosis and the generation of free radicals may be involved in the pathogenesis of systemic sclerosis.
In systemic sclerosis, affected organs and systems include the skin, lungs, heart, digestive system, kidneys, muscles, joints, and nervous system.
Etiology
Systemic sclerosis is an autoimmunologic disease, but the pathogenesis is only partially understood. Certain factors are well known to trigger occurrence of the disease or create a similar clinical appearance. Environmental factors include exposure to the following:
-
Vibration injury (similar vascular changes)
-
Organic solvents (eg, toluene, benzene, xylene)
-
Aliphatic hydrocarbons (eg, hexane, vinyl chloride, trichloroethylene)
-
Epoxy resin
-
Amino acid compound L-5-hydroxytryptophan
-
Pesticides
-
Drugs (eg, bleomycin, carbidopa, pentazocine, cocaine, penicillamine, vitamin K): A limited form of cutaneous systemic sclerosis has been described with paclitaxel in with the setting of breast cancer. [5]
-
Appetite suppressants (eg, phenylethylamine derivatives)
-
Substances used in cosmetic procedures (eg, silicone or paraffin implants)
Epidemiology
Frequency
A 2019 epidemiology study reported that based on 39 publications, the prevalence of systemic sclerosis in Europe and North America was 7.2-33.9 cases per 100,000 individuals, with an annual incidence rate of 0.6-2.3 cases per 100,000 individuals. [6]
Systemic sclerosis is rare in the resident population of Japan and China.
Race
No apparent racial predominance exists. However, systemic sclerosis is rare in the resident population of Japan and China. Diffuse systemic sclerosis (dSSc) occurs more often in black women than in white women.
Sex
Overall, a substantial female predominance exists, with a female-to-male ratio of 3-6:1. However, dSSc occurs equally in males and females. The limited form of systemic sclerosis (lSSc) has a strong female predominance, with a female-to-male ratio of 10:1. Another analysis showed that men tend to have diffuse disease and women to have calcinosis. [7]
Age
Systemic sclerosis usually appears in women aged 30-40 years, and it occurs in slightly older men. In approximately 85% of cases, systemic sclerosis develops in individuals aged 20-60 years. Cases also are observed in children and in the elderly population.
Prognosis
The prognosis depends on the type of systemic sclerosis (SSc). In lSSc, a patient's condition can be stable for years. However, in dSSc, the disease can rapidly lead to death, if it is not treated promptly. Pulmonary hypertension may be an important cause of mortality in these patients. Survival complicated by pulmonary hypertension remains poor despite currently available treatment options. [8] Pulmonary hypertension was documented 25% of elderly patients, yet only in 12% of the youngest ones in a large study that stressed the differences in early- versus late-onset systemic sclerosis. [9] Cigarette smoking has been found to reduce overall survival. [10] In a multicenter French cohort study, age older than 60 years at diagnosis, diffuse cutaneous SSc, scleroderma renal crisis, dyspnea, 6-minute walking distance, forced vital capacity less than 70%, diffusing capacity of the lungs for carbon monoxide less than 70%, pulmonary hypertension, telangiectasia, valvular disease, malignancy, anemia, and C-reactive protein greater than 8mg/L were linked with poorer survival. [11]
The undeniable impact of systemic sclerosis on quality of life underscores the need for a biopsychosocial approach to the clinical management. Orofacial manifestations need to be considered. [12] Timely detection of psychosocial difficulties and appropriate psychological or psychiatric intervention are also important steps toward better adherence to medical treatment. [13] Not surprisingly, the prevalence of depressive symptoms were evaluated as high and were independently associated with pain, fatigue, social support, emotion-focused coping, helplessness, and fear of progression. [14, 15] Psychosocial factors associated with depressive symptoms merit attention.
A Canadian study assessed the World Health Organization Disability Assessment Schedule II (WHODAS II) as a valid measure of quality of life in systemic sclerosis patients. Results suggested the WHODAS II has good psychometric properties and is a valid measure of health-related quality of life in patients with systemic sclerosis. [16]
Mortality/morbidity
The mortality rate is increasing in the United States and Europe; as many as 3.08 persons are affected per 1 million. Generally, renal and lung changes are responsible for death in patients with systemic sclerosis. Pulmonary hypertension leads to 12% of systemic sclerosis–related deaths. Lung fibrosis and heart changes are responsible for 9% of systemic sclerosis–related deaths. These patients also have an increased risk of venous thromboembolism. [17]
Patient Education
The most effective method for preventing a Raynaud episode is to avoid exposure to cold temperatures. Smoking cessation should be discussed with patients and their families, as applicable.
-
Face of 65-year old woman with systemic sclerosis and skin thickening of 20 years' duration: Note the pinched nose, taut skin with numerous telangiectasias, and retraction of the lips.
-
Telangiectasias affecting the face: They are pronounced and numerous, especially in the atrophic phase of the disease. Radical furrowing around the mouth is also characteristic in the later stage of the disease.
-
Raynaud phenomenon of the hands: Symmetrical acral vasospasm is present, with characteristic pallor, cyanosis, suffusion, and a sense of fullness and tautness.
-
Puffy appearance of the woman's hand in the edematous phase of early scleroderma.
-
In systemic sclerosis, ulceration at the tip of the finger is regarded to be secondary to ischemia.
-
Hand of a woman with scleroderma of several years' duration: The thickened, tight, thin skin over the fingers is the result of self-amputation of the distal phalanx due to ischemia. Moderately severe flexion contractures of the fingers are present.
-
In systemic sclerosis, skin hyperpigmentation of the lower legs is surrounded by areas of hypopigmentation. The result is a salt-and-pepper appearance.
Tables

What would you like to print?
- Addressing Orofacial Changes in Scleroderma
- The Hidden Impact of Systemic Sclerosis Without Scleroderma
- Multiple Common Comorbidities Can Have Big Impact on Systemic Sclerosis Outcomes
-
 Joint Pain in Paradise: A Closer Look at Arthritis and HS
Joint Pain in Paradise: A Closer Look at Arthritis and HS
-
Potassium-Competitive Acid Blockers (PCABs) Provide a New Weapon to Combat Erosive Esophagitis
-
How Patient-Centered Research Is Transforming Bipolar Disorder Care and Dispelling Stigma





