Practice Essentials
Apert syndrome is a rare autosomal dominant disorder characterized by craniosynostosis, craniofacial anomalies, and severe symmetrical syndactyly (cutaneous and bony fusion) of the hands and feet (see the images below). It is probably the most familiar and best-described type of acrocephalosyndactyly. Reproductive fitness is low, and more than 98% of cases arise by new mutation.The syndrome is named for the French physician who described the syndrome acrocephalosyndactylia in 1906. [1]
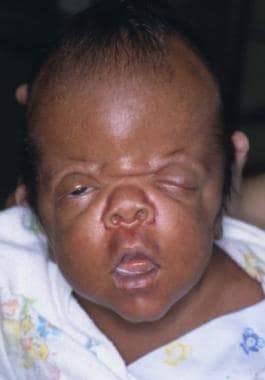 An infant with Apert syndrome is shown. Note the characteristic ocular hypertelorism, down-slanting palpebral fissures, proptotic eyes, horizontal groove above the supraorbital ridge, break of the eyebrows' continuity, depressed nasal bridge, and short, wide nose with bulbous tip.
An infant with Apert syndrome is shown. Note the characteristic ocular hypertelorism, down-slanting palpebral fissures, proptotic eyes, horizontal groove above the supraorbital ridge, break of the eyebrows' continuity, depressed nasal bridge, and short, wide nose with bulbous tip.
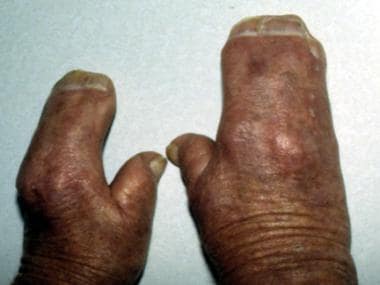 Note the mitten appearance of the hands with syndactyly involving the second, third, fourth, and fifth fingers. This patient also has characteristic concave palms, hitchhiker posture (radial deviation) of the short broad thumbs, and contiguous nail beds (synonychia).
Note the mitten appearance of the hands with syndactyly involving the second, third, fourth, and fifth fingers. This patient also has characteristic concave palms, hitchhiker posture (radial deviation) of the short broad thumbs, and contiguous nail beds (synonychia).
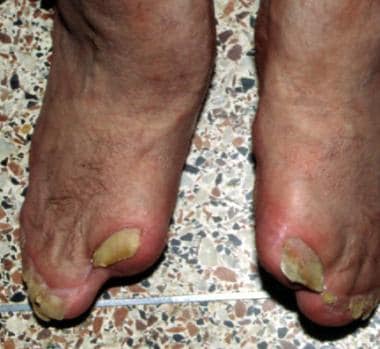 Note the socklike appearance of the feet with syndactyly involving the second, third, fourth, and fifth toes. The patient also has contiguous nail beds (synonychia).
Note the socklike appearance of the feet with syndactyly involving the second, third, fourth, and fifth toes. The patient also has contiguous nail beds (synonychia).
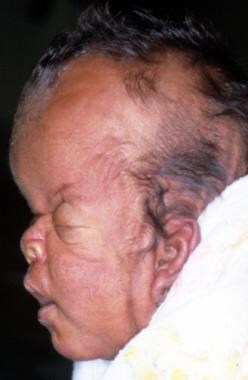 In this profile photo, turribrachycephaly (high prominent forehead), proptosis, a depressed nasal bridge, a short nose, and low-set ears are prominent.
In this profile photo, turribrachycephaly (high prominent forehead), proptosis, a depressed nasal bridge, a short nose, and low-set ears are prominent.
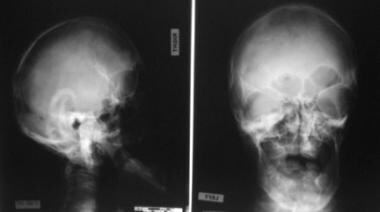 This radiograph demonstrates turribrachycephaly, shallow orbits, ocular hypertelorism, and a hypoplastic maxilla.
This radiograph demonstrates turribrachycephaly, shallow orbits, ocular hypertelorism, and a hypoplastic maxilla.
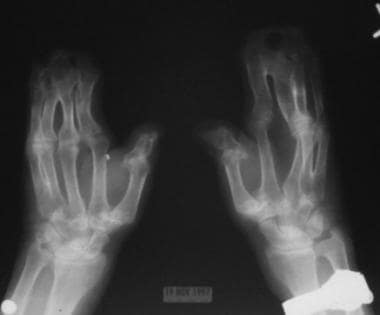 Note the osseous syndactyly involving the second, third, fourth, and fifth fingers; multiple synostosis involving the distal phalanges and proximal fourth and fifth metacarpals; symphalangism of the interphalangeal joints; shortening and radial deviation of the distal phalanx; and the delta-shaped deformity of proximal phalanx of the thumbs.
Note the osseous syndactyly involving the second, third, fourth, and fifth fingers; multiple synostosis involving the distal phalanges and proximal fourth and fifth metacarpals; symphalangism of the interphalangeal joints; shortening and radial deviation of the distal phalanx; and the delta-shaped deformity of proximal phalanx of the thumbs.
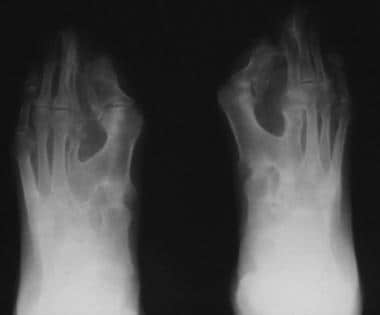 Note the osseous syndactyly, fusion of the interphalangeal joints, synostosis involving the proximal first and second metatarsals, and the partially duplicated and delta-shaped proximal phalanx of the great toes.
Note the osseous syndactyly, fusion of the interphalangeal joints, synostosis involving the proximal first and second metatarsals, and the partially duplicated and delta-shaped proximal phalanx of the great toes.
Signs and symptoms of Apert syndrome
With craniosynostosis, coronal sutures most commonly are involved, resulting in acrocephaly, brachycephaly, turribrachycephaly, flat occiput, and high prominent forehead.
Patients have apparent low-set ears, with occasional conductive hearing loss and congenital fixation of the stapedial footplate.
The eyes exhibit down-slanting palpebral fissures, hypertelorism, shallow orbits, proptosis, exophthalmos, strabismus, amblyopia, optic atrophy, and, rarely, luxation of the eye globes, keratoconus, ectopic lentis, congenital glaucoma, lack of pigment in the fundi with occasional papilledema, and preventable vision loss or blindness.
The nose has a markedly depressed nasal bridge. It is short and wide, with a bulbous tip, parrot-beaked appearance, and choanal stenosis or atresia.
The mouth area has a prominent mandible, down-turned corners, a high arched palate, a bifid uvula, and a cleft palate.
The upper limbs are more severely affected than lower limbs. Coalition of distal phalanges and synonychia found in the hands are never present in the feet. The glenohumeral joint and proximal humerus are more severely affected than the pelvic girdle and femur. The elbow is much less severely involved than the proximal portion of the upper limb.
Intelligence varies in persons with Apert syndrome from normal to mental deficiency, although a significant number of patients have intellectual disability.
Apert syndrome is also characterized by cutaneous, cardiovascular, genitourinary, gastrointestinal, and respiratory disorders.
Workup in Apert syndrome
Regarding the molecular analysis of Apert syndrome, it is known that more than 98% of cases are caused by specific missense substitution mutations involving adjacent amino acids (Ser252Trp, Ser252Phe, or Pro253Arg) in exon 7 of FGFR2.
Imaging studies in Apert syndrome include the following:
-
Skull radiography
-
Spinal radiography
-
Limb radiography
-
Hand radiography
-
Foot radiography
-
Computed tomography (CT) scanning - CT scanning with comparative three-dimensional reconstruction analysis of the calvaria and cranial bases has become the most useful radiologic examination in identifying skull shape and the presence or absence of involved sutures
-
Magnetic resonance imaging (MRI) - MRI reveals the anatomy of the soft-tissue structures and associated brain abnormalities (ie, nonprogressive ventriculomegaly; hydrocephalus; complete or partial absence of the septum pellucidum; absence of septal leaflets; and thinning, deficiency, or agenesis of the corpus callosum) [2, 3]
Management of Apert syndrome
Surgical management of Apert syndrome can include the following:
-
In severe cases, lateral or medial tarsorrhaphy to protect the corneas
-
In rare cases, orotracheal intubation to manage upper airway obstruction during the neonatal period
-
Tracheostomy in children severely affected by sleep apnea
-
Bilateral myringotomy and placement of ventilation tubes to manage chronic middle ear effusion associated with bilateral conductive hearing deficit
-
Cranial surgery to remove synostotic sutures; reshape the calvaria; allow more normal cranial development to proceed with respect to shape, volume, and bone quality; and relieve increased intracranial pressure
-
Orbital surgery to correct ocular proptosis, reduce increased interorbital distance (hypertelorism), and correct increased interior malrotation
-
Nasal surgery to correct the excessively obtuse nasofrontal angle, flat nasal dorsum, and ptotic nasal tip, in infants and children, and to reduce the nasal tip bulk in teenagers and adults
-
Midfacial surgery to normalize midface appearance, expand the inferior orbit, volumetrically expand the nasal and nasopharyngeal airways, and establish a normal dentoskeletal relationship
-
Mandibular osteotomy to improve dentoskeletal relations for masticatory and aesthetic benefit
Pathophysiology
During early infancy (< 3 mo), the coronal suture area is prematurely closed. A bony condensation line beginning at the cranial base and extending upward with a characteristic posterior convexity represents this occurrence. Anterior and posterior fontanelles are widely patent. The midline of the calvaria has a gaping defect, extending from the glabellar area to the posterior fontanelle via the metopic suture area, anterior fontanelle, and sagittal suture area. The skull with a gaping midline defect appears to permit adequate accommodation of the growing brain. The lambdoidal sutures appear normal in all cases.
During the first 2-4 years of life, the midline defect is obliterated by coalescence of the enlarging bony islands without evidence of any proper formation of sutures. An extreme short squama and orbital part of the frontal bone together with the posterior convexity of the coronal bone condensation line suggest that growth inhibition in the sphenofrontal and coronal suture area has its onset very early in fetal life.
Unique fibroblast growth factor receptor 2 (FGFR2) mutations lead to an increase in the number of precursor cells that enter the osteogenic pathway. Ultimately, this leads to increased subperiosteal bone matrix formation and premature calvaria ossification during fetal development. The order and rate of suture fusion determine the degree of deformity and disability. Once a suture becomes fused, growth perpendicular to that suture becomes restricted, and the fused bones act as a single bony structure. Compensatory growth occurs at the remaining open sutures to allow continued brain growth; however, complex, multiple sutural synostosis frequently extends to premature fusion of the sutures at the base of the skull, causing midfacial hypoplasia, shallow orbits, a foreshortened nasal dorsum, maxillary hypoplasia, and occasional upper airway obstruction.
A retrospective study by Kolar et al examined the characteristics of mandibular growth associated with FGFR2 mutations, including in children with Apert, Crouzon, or Pfeiffer syndrome, with initial measurements finding slightly greater than normal mandibular height and bigonial breadth and deficient sagittal depth and cranial base width. Slight early growth acceleration was found along the vertical and sagittal axes, while growth was deficient at the cranial base. The mature skeleton was marked by above average mandibular vertical height and bigonial width, with deficiency still found in the mandibular depth (forward sagittal growth) and cranial base width. [4]
The first genetic evidence that syndactyly in Apert syndrome is a keratinocyte growth factor receptor (KGFR)-mediated effect was provided by the observation of the correlation between KGFR expression in fibroblasts and severity of syndactyly. Patients with Ser252Trp and those with Pro253Arg have different phenotypic expression. The syndactyly is more severe with Pro253Arg mutation for both hands and feet, whereas cleft palate is significantly more common with Ser252Trp mutation. [5]
Amblyopia and strabismus are more common in patients with the FGFR2 Ser252Trp mutation, and optic disc pallor is more frequent in patients with the FGFR2 Pro253Arg mutation. [6] Patients with FGR2 Ser252Trp mutations have a significantly greater prevalence of visual impairment compared with patients with the FGFR2 Pro253Arg mutation. [6, 7]
Epidemiology
Frequency
United States
-
Apert syndrome accounts for 4.5% of all cases of craniosynostosis.
Mortality/Morbidity
See the list below:
-
Most patients experience some degree of upper airway obstruction during infancy. Upper airway compromise due to reduction in nasopharynx size and choanal patency as well as lower airway compromise due to anomalies of the tracheal cartilage may be responsible for early death.
-
Sleep apnea syndrome is common. Upper airway compromise, consisting of obstructive sleep apnea and cor pulmonale, may result from small nasopharyngeal and oropharyngeal dimension in the Apert craniofacial configuration.
-
Patients are at risk for complications resulting from elevated intracranial pressure despite surgical attempts to increase cranial capacity in infancy.
Race
See the list below:
-
Asians have the highest prevalence (22.3 cases per million live births).
-
Hispanics have the lowest prevalence (7.6 cases per million live births).
Sex
See the list below:
-
Apert syndrome has no sex predilection.
Age
See the list below:
-
Apert syndrome is detected in the newborn period due to craniosynostosis and associated findings of syndactyly in the hands and feet.
-
An infant with Apert syndrome is shown. Note the characteristic ocular hypertelorism, down-slanting palpebral fissures, proptotic eyes, horizontal groove above the supraorbital ridge, break of the eyebrows' continuity, depressed nasal bridge, and short, wide nose with bulbous tip.
-
Note the mitten appearance of the hands with syndactyly involving the second, third, fourth, and fifth fingers. This patient also has characteristic concave palms, hitchhiker posture (radial deviation) of the short broad thumbs, and contiguous nail beds (synonychia).
-
Note the socklike appearance of the feet with syndactyly involving the second, third, fourth, and fifth toes. The patient also has contiguous nail beds (synonychia).
-
In this profile photo, turribrachycephaly (high prominent forehead), proptosis, a depressed nasal bridge, a short nose, and low-set ears are prominent.
-
This radiograph demonstrates turribrachycephaly, shallow orbits, ocular hypertelorism, and a hypoplastic maxilla.
-
Note the osseous syndactyly involving the second, third, fourth, and fifth fingers; multiple synostosis involving the distal phalanges and proximal fourth and fifth metacarpals; symphalangism of the interphalangeal joints; shortening and radial deviation of the distal phalanx; and the delta-shaped deformity of proximal phalanx of the thumbs.
-
Note the osseous syndactyly, fusion of the interphalangeal joints, synostosis involving the proximal first and second metatarsals, and the partially duplicated and delta-shaped proximal phalanx of the great toes.
-
A 9-month-old girl was seen because of syndactyly of the hands and feet as well as associated with craniofacial anomalies. The family and pregnancy histories were noncontributory. The child had broad thumbs with 2-5 digits with cutaneous syndactyly (only the right hand is shown here). The feet were characterized by brachydactyly and syndactyly of 2-5 toes. Genomic DNA analysis showed a heterozygous C-to-G mutation at nucleotide 755 of the fibroblast growth factor receptor 2 (FGFR2) gene (c.755C>G) that changes a codon for serine (TCG) to that for tryptophan (TGG) at amino acid position 252 (p.Ser252Trp). This mutation is diagnostic for Apert syndrome.
-
The right hand radiograph for the same patient in the previous image, at age 15 months (left image), showed soft-tissue fusion between the second through fourth digits as well as fusion of the proximal soft tissues between the fourth and fifth digits. Hypoplastic, deformed phalanges were present with fusion of the proximal and middle phalanges of the second through fourth digits. Bony fusion was also seen at the bases of the fourth and fifth metacarpals along with fusion of the capitate and hamate. The thumb pointed laterally with a sharp angulation at the first metacarpophalangeal joint. A right hand radiograph from the child at age 1 month (right image) is provided for comparison. Similar abnormalities were also seen in the left hand (not shown).
-
Radiographs of both feet in the same child as in the previous images, at age 1 month, showed foreshortening of the bilateral second metatarsals, the right third proximal phalanx and left fourth phalanx, and the distal phalanges of the left second, third, fourth, and fifth digits. Both great toes are bulbous and foreshortened, with deformed phalanges and partially duplicated metatarsals. Soft-tissue fusion was present in the second through fifth digits of both feet.
-
Magnetic resonance images of the brain obtained at in the same patient as in the previous images, at age 16 months, showed hypoplasia of the parieto-occipital white matter, with undulating bilateral lateral ventricle occipital horns (arrow; left image). Shallow orbits can be appreciated bilaterally with ocular hypertelorism (right image).
-
A 17-month-old boy with Apert syndrome. Three-dimensional (3-D) CT imaging of the head showed craniosynostosis of the coronal suture and a hypoplastic midface.
-
Axial facial CT scans in the same patient as in the previous image showed bilateral shallowed orbits with proptosis and mild hypertelorism. Crowding of the teeth was present.
-
Radiograph of bilateral hands in the same patient as in the previous images, at age 18 months, showed bilateral syndactyly involving the second through fifth digits, with absent distal phalanges. Bilateral shortened hypoplastic middle phalanges are present, with bony fusion at the third and fourth middle phalanges, along with deformities of the proximal phalanges and shortened metacarpals.
-
Radiograph of bilateral feet in the same patient as in the previous images, at age 7 years, showed syndactyly with diffuse deformities and multiple midfoot and hindfoot tarsal coalitions.
-
Three-dimensional facial CT scans in the same child as in the previous images, at age 9 years, showed hypoplastic midfacial bones.
