Practice Essentials
Teratomas (from Greek teras, "monster," and -oma, a suffix denoting a tumor or neoplasm) and other germ cell tumors (GCTs) are relatively common solid neoplasms in children. They may occur in both gonadal and extragonadal locations. Locations and specific tumor types depend on the age of the child. These tumors are grouped together because they all appear to arise from postmeiotic germ cells. Most of the malignant tumors produce markers that can be serologically assessed. [1, 2, 3, 4, 5, 6, 7, 8]
For children with extracranial GCTs, surgery is an essential component of treatment. Depending on the clinical factors present, appropriate treatment may involve one of the following:
-
Surgical resection followed by careful monitoring for disease recurrence
-
Initial surgical resection followed by platinum-based chemotherapy
-
Diagnostic tumor biopsy and preoperative platinum-based chemotherapy followed by definitive tumor resection
Pathophysiology
Several theories about the origin of these tumors have been advanced. The best evidence suggests that most are due to abnormal differentiation of fetal germ cells that arise from the fetal yolk sac. Normal migration of these germ cells may cause gonadal tumors, whereas abnormal migration produces extragonadal tumors. [9] Teratomas are typically found in the midline or gonads. Frequencies of the most common sites are as follows:
-
Sacrococcygeal - 40%
-
Ovary - 25%
-
Testicle - 12%
-
Brain - 5%
-
Other (including the neck and mediastinum) - 18%
By definition, teratomas include components derived from all three embryonic layers: ectoderm, endoderm, and mesoderm. These tissues are foreign to the location in which they are found.
Teratomas may be classified as mature or immature on the basis of the presence of immature neuroectodermal elements within the tumor. Mature tumors (grade 0) have no immature elements. In grade 1 tumors, immature elements are limited to one low-power field per slide; in grade 2 tumors, fewer than four fields are present per slide; and in grade 3 tumors, more than four fields are present per slide.
In the past, survival was linked to the degree of immaturity in the teratoma. Close histologic evaluation of immature teratomas reveals a good correlation between the degree of immaturity and the presence of microscopic foci of frankly malignant elements. These malignant elements are typically yolk sac tumors but may also represent primitive neuroectodermal tumors (PNETs). Charoenkwan et al found overexpression of p53 in the more aggressive immature teratomas at all sites. [10]
The risk of recurrence also appears to be related to the degree of immaturity. Recurrence in a completely resected mature teratoma is less than 10%; in an immature teratoma, recurrence may be as high as 33%.
The likelihood of recurrence depends on the site of the tumor as well as the completeness of resection. The German MAKEI trials suggested that the recurrence rate for immature teratomas could be decreased to 9.5% with chemotherapy. [11] Sacrococcygeal teratomas are more likely to recur than those in the ovary or other sites. Molecular biologic and cytogenetic studies are providing a firmer scientific basis for these observations.
In 1965, Teilum first suggested the germ cell origin of gonadal tumors. [12] Since that time, the pathologic classification scheme has evolved to its present state. Germ cells undergo neoplastic transformation as follows:
-
Suppressed differentiation - Seminoma; dysgerminoma
-
Differentiation - Initial (embryonal carcinoma); embryonic (mature teratoma and immature teratoma); extraembryonic (choriocarcinoma and endodermal sinus tumor [yolk sac tumor])
Mutter described genetic imprinting as a major factor in the development of some of these tumors. [13] The developmentally expressed genes for insulinlike growth factor 2 (IGF II) and its receptor RNA (H-19); small nuclear riboprotein (SNRPN); mas proto-oncogene; and the tumor suppressor genes WT1 and MASH2 are imprinted, depending on their maternal or paternal origin.
Mutter suggested that these genes or the cells have only the maternal imprint because many teratomas arise from a parthenogenetically activated egg. Therefore, maternally active genes are present in higher than usual concentrations, and maternally inactive products are present at lesser concentrations if at all. These abnormalities may account for the lack of organization of the three germ cell layers.
Oosterhuis et al suggested that tumors may be grouped on the basis of their chromosomal abnormalities as follows [14] :
-
Group 1, including immature teratomas and yolk sac tumors - Immature teratomas are usually diploid, whereas yolk sac tumors may be diploid, tetraploid, or aneuploidy; chromosomal aberrations include overrepresentation of chromosomes X, 1, 3, 8, 12, and 14 and underrepresentation of Y and X; deletions in 1p and rearrangements of 3q and 6q may be present; isochromosome 12p (i12p) has been found; an abnormal number of centromeres is frequent in both diploid and aneuploid tumors
-
Group 2, including most nonseminomatous malignant GCTs and typically including numeric abnormalities in X, 7, 8, 12, and 21 as excess and deletions of Y, 11, 13, or 18 - Once again, isochromosomes 12p with other aberrations of 12p and 1p are present
-
Group 3, including mature teratomas or mature cystic teratomas - Numeric abnormalities, including extra X, 7, 12, and 15, have been found; no chromosomal structural anomalies have been found
-
Group 4, including spermatocytic seminoma, a type usually confined to older men - The cytogenetics of this group have not been characterized; as with abnormalities and imprinting patterns, these chromosomal rearrangements can lead to overproduction of certain gene products and underproduction of others; these lead to the abnormal growth characteristics of the tumor
Hara et al suggested that the MAGE gene family of tumor rejection antigens may also be involved in the pathogenesis of these tumors. [15] These genes appear to be more active in pure seminoma or mixed type of seminomatous elements than in other GCTs. In their limited study of 22 patients, MAGE expression was not correlated with disease progression. It is likely to be only an indicator of maturity or differentiation of the tissues.
The concept of teratoma with malignant transformation indicates the development of non–germ cell malignancies within a teratoma. Among 641 patients in the MAKEI protocols 83/86/89/96, nine patients were identified with this finding. [16] Five patients presented with a carcinoma, two with glial tumors, and two with embryonal tumors. Resection and chemotherapy were typically used. Because these tumors are quite rare, response to treatment is difficult to generalize. [17]
When tumors resistant to platinum-based chemotherapy are evaluated, between one third and one half of them exhibit microsatellite instability (MSI).
Etiology
With sacrococcygeal teratomas, no causative agents are known. With respect to ovarian GCTs, a familial predilection may be present. Cases in seven families have been reported in which female first-degree relatives had GCTs. In an additional seven families, males had GCTs. This observation suggests that certain genes may be present in these families, predisposing them to germ cell malignancy.
One study that examined the effect of diet on the development of ovarian tumors revealed that diets high in polyunsaturated fat were associated with the development of teratomas. [18] It is likely that plant estrogens, and not the polyunsaturated fat, are associated with an increased tumor risk.
It has been suggested that cryptorchidism increases the risk of GCT by a factor of 10.8. [19] Tumors may appear in the ipsilateral or contralateral testicle. Hernia is similarly associated with GCTs. One study also revealed that a history of pyloric stenosis leads to a fourfold risk of germ cell malignancy. [20]
Boys whose father or brother has had a teratoma have a 5-15% increased risk for teratoma. Whether this increased risk is due to genetic causes or is a consequence of shared environment remains to be defined. [21]
No major high-penetrance susceptibility gene for testicular GCTs has been identified; inheritance is likely driven by a complex polygenic model. [22] The most common genomic alterations associated with these tumors include gains in chromosome 12p and mutations in KIT, KRAS, and NRAS, particularly in seminomas.
Differences (disorders) of sex development (DSDs) have also been associated with development of GCTs. Gonadoblastoma is observed in roughly one third of patients with DSDs. Although gonadoblastoma is a carcinoma in situ, it frequently evolves into dysgerminoma; yolk sac tumors, immature teratomas, and choriocarcinomas are possible as well. [23]
Turner syndrome is similarly a risk factor for gonadoblastoma. Klinefelter syndrome has been linked with an increased risk of extragonadal malignant GCTs. [24] The highest risk seems to be among patients who carry some Y-chromosome genes in ectopic locations where they may not be normally regulated.
Children with DSDs typically have 46,XY DSD (previously referred to as male pseudohermaphrodites) with antigen insensitivity or 5-alpha reductase deficiency. These patients with testicular feminization are sometimes discovered serendipitously during a hernia repair.
Optimal timing for gonadal resection in these situations is a matter of debate. Gonadal estrogen production may benefit the patient in terms of growth and development. However, gonadoblastoma has been observed in patients as young as 2 months, and frank tumors have been observed in those younger than 2 years. The decision to leave or remove the gonads early should be made with the family after thorough discussion of these risks and potential benefits.
Epidemiology
United States statistics
Sacrococcygeal teratoma occurs in 1 in 30,000-70,000 live births. Ovarian teratomas are almost as common, whereas testicular teratomas are about one third less frequent. The overall incidence of malignant GCTs is approximately 3% of all childhood malignancies, or approximately 3 cases per million population per year. The frequency of all GCTs has increased over the past several decades.
International statistics
A study assessing the global incidence of ovarian GCTs in three age groups (0-9 y, 10-19 y, 20-39 y) found that the highest frequency was in 10- to 19-year-olds and that the incidence was generally highest in Eastern Asia, Central America, and North America. [25] Less geographic variation in incidence was noted in 0- to 9-year-olds than in the other two groups. Overall, the highest incidence figures and the largest increases in incidence were seen in Eastern Asia.
Age-related demographics
Sacrococcygeal teratomas
Sacrococcygeal teratomas are congenital. Those with a significant external component (see the image below) are identified at birth. Tumors without an external component (Altman type 4) are discovered later. When the tumors are resected before the patient is aged 2 months, 7-10% are malignant. After that age, the risk of malignancy increases greatly, exceeding 50% by age 1 year.
 Sacrococcygeal teratoma in a female neonate. This particular tumor is largely external with no intrapelvic extension.
Sacrococcygeal teratoma in a female neonate. This particular tumor is largely external with no intrapelvic extension.
Ovarian tumors
The incidence of ovarian GCTs (see the image below) increases with age and peaks around age 15-19 years. When girls younger than 15 years were examined, fewer than 10% of tumors occurred in girls younger than 5 years, 20% were found in girls aged 5-9 years, and more than 70% were found in girls aged 10-14 years. [26]
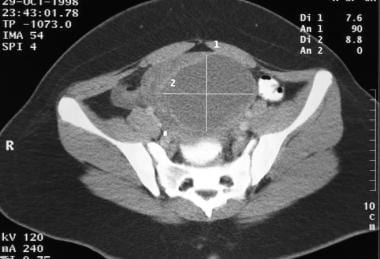 This is an ovarian mixed germ cell tumor in a 13-year-old girl. This tumor caused right lower quadrant pain. It is largely cystic in composition. No calcifications are observed within the mass.
This is an ovarian mixed germ cell tumor in a 13-year-old girl. This tumor caused right lower quadrant pain. It is largely cystic in composition. No calcifications are observed within the mass.
Benign ovarian tumors, largely teratomas, predominate. Roughly 70% of malignant ovarian tumors in childhood are GCTs, one quarter are epithelial, and the remainder are stromal tumors. The ratio of GCTs to epithelial malignancies decreases with increasing patient age.
Chromosomal abnormalities also appear to be related to age at presentation for teratomas. In girls younger than 5 years, no chromosomal abnormalities were found, whereas older girls often have gains of 12p and chromosomes 7 and 8.
Testicular tumors
Testicular GCTs in childhood are split between teratomas and yolk sac tumors (see the image below). They are more common from birth to age 5 years. From age 6 years until puberty, testicular tumors are exceedingly uncommon. Thereafter, the incidence increases, and a more adultlike tumor pattern develops, with seminomas gradually becoming the predominant histology.
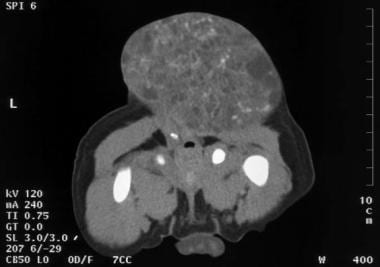
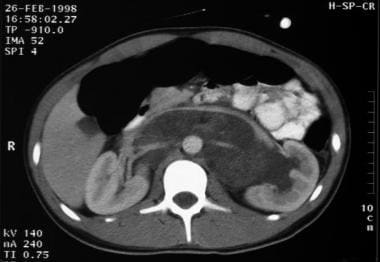 Yolk sac tumor of the testis. The tumor is metastatic to the retroperitoneum. It encases the aorta and renal arteries. The vena cava and renal veins are displaced anteriorly by the mass.
Yolk sac tumor of the testis. The tumor is metastatic to the retroperitoneum. It encases the aorta and renal arteries. The vena cava and renal veins are displaced anteriorly by the mass.
Both teratomas and yolk sac tumors may be associated with contralateral in-situ dysgenesis in 9% of patients compared with 0.5% of otherwise healthy males. Contralateral tumors are often found. These are occasionally synchronous but are more often metachronous. Ongoing surveillance of the contralateral testis is therefore needed.
Among malignant GCTs, yolk sac tumors predominate until the age of 14 years. Few tumors of any type are diagnosed in children aged 5-9 years. For malignant teratomas, yolk sac tumors, and all GCTs, respective rates by age group are as follows:
-
Age group 0-4 years - 0.45 case, 3.66 cases, and 4.16 cases per million population
-
Age group 5-9 years - 0.12 case, 0.12 case, and 0.12 case per million population
-
Age group 10-14 years - 0.05 case, 1.30 case, and 1.77 case per million population
Sex-related demographics
Sacrococcygeal teratoma has a 4:1 female-to-male predominance. In other GCTs, the female-to-male ratio is roughly 2:1 in children.
Race-related demographics
Overall racial predispositions for these tumors have not been established. However, a population-based case-control study using figures from the California Cancer Registry, which included 451 cases of malignant GCTs in children aged 0-5 years, found that Asian and Pacific Islander children were at higher risk for developing these tumors than non-Hispanic White children were. [27]
Prognosis
Mortality for congenital teratomas depends on gestational age and the size and location of the tumors. Survival of preterm infants younger than 30 weeks' gestation with sacrococcygeal teratoma is only 7%, whereas the survival for infants older than 30 weeks' gestation is 75%.
Rapid early growth is associated with the yolk-sac phenotype and carries a poorer prognosis. [28] Early tumors are frequently large in relation to the size of the infant and may induce congestive heart failure. Cervical teratomas may frequently lead to airway problems and death when they are large.
Before current developments in chemotherapy, the 10-year survival rate for malignant GCTs ranged from 25% for embryonal carcinoma to 75% for dysgerminoma. Today, overall survival rates are higher than 90%. However, subpopulations exist that have worse event-free survival (EFS) and overall survival (OS) rates. [29]
Now that survival of most patients is approaching 100%, the morbidity of treatment, particularly with respect to fertility, [30] must be addressed. Because these patients are often prepubertal, standard means of collecting and preserving sperm or ova are not applicable. Techniques for preserving normal gonadal tissue and maturing it to produce viable sperm and ova must be developed.
Long-term follow-up of these patients is necessary because of the possibility of chronic pulmonary disease, hearing loss, and other long-term toxicities of the chemotherapy used in treating these lesions.
In a single-center retrospective study evaluating long-term outcomes in 25 pediatric patients with a primary mediastinal GCT, six patients were treated with resection alone and were cured without disease recurrence or progression, whereas 19 were treated for malignancy and had a 5-year overall survival of 0.39 ± 0.12. [31] Localized disease, complete resection, and platinum-based chemotherapy were identified as factors linked with improved survival in malignant nonseminomatous mediastinal GCTs, and neoadjuvant platinum-based three-drug therapy followed by delayed surgical resection was found to be the appropriate treatment modality for these tumors.
Pediatric Intergroup Study findings
Intermediate risk
The results of the 2004 Intergroup GCT study showed excellent EFS and OS. [32, 33]
Treatment with four cycles of standard-dose cisplatin-etoposide-bleomycin therapy resulted in 6-year EFS of 95% and OS of 95.7%. Results for specific tumors and stages included the following:
-
Stage II testicular tumors - 100% EFS and 100% OS
-
Stage I ovarian tumors - 95.1% EFS and 95.1% OS
-
Stage II ovarian tumors - 87.5% EFS and 93.8% OS
Two patients died of recurrent disease, and one patient died of secondary acute myelocytic leukemia. Toxicity was limited with this drug regimen; occasional low-grade toxicity to the renal, pulmonary, and auditory systems were reported. More severe hematologic toxicities were reported.
High risk
The 2004 Intergroup high-risk germ cell trial compared standard-dose cisplatin-etoposide-bleomycin treatment with high-dose treatment. [32, 33] Although substantial improvement in EFS was noted in the high-dose arm, OS was not significantly improved, and serious renal toxicity (7% reduced creatinine clearance) and ototoxicity (14% testable hearing loss) were frequent.
Survival rates were as follows:
-
High-dose - 89.6% EFS and 91.7% OS
-
Standard dose - 80.5% EFS and 86.0% OS
-
Gonadal stage III tumors - 94-96% EFS and 98-100% OS
-
Gonadal stage IV tumors - 86-88% EFS and 90-93% OS
-
Extragonadal stage I and II tumors - 89% EFS and 93% OS
-
Extragonadal stage III and IV tumors - 75-78% EFS and 81% OS
Because the high-stage extragonadal GCTs had substantially reduced survival compared with all other groups, more aggressive therapy may be warranted in those patients.
-
Sacrococcygeal teratoma in a female neonate. This particular tumor is largely external with no intrapelvic extension.
-
This is an ovarian mixed germ cell tumor in a 13-year-old girl. This tumor caused right lower quadrant pain. It is largely cystic in composition. No calcifications are observed within the mass.
-
Ovarian yolk sac tumor at surgery.
-
Yolk sac tumor of the testis. The tumor is metastatic to the retroperitoneum. It encases the aorta and renal arteries. The vena cava and renal veins are displaced anteriorly by the mass.
-
Esophagram in an infant with massive thoracic germ cell tumor. Note how the esophagus is displaced posteriorly and laterally by the left mediastinal tumor.
-
Chest radiograph of the patient in Media file 5 after treatment with chemotherapy. The size of the tumor has not decreased.
-
CT scan of the chest in the patient in Media file 6. The carina is displaced posteriorly and to the right. The vena cava is displaced anteriorly, and the aorta is compressed between the mass and the spine.
Tables
| Risk | Site | Stage | Age |
|---|---|---|---|
| Low | Testis | I | Any |
| Ovary | I | Any | |
| Extragonadal | I | Any | |
| Standard 1 | Testis | II-IV | < 11 y |
| Ovary | II-IV | < 11 y | |
| Extragonadal | II-IV | < 11 y | |
| Standard 2 | Testis (IGCCC good risk) | II-IV | >11 y |
| Ovary | II-IV | >11 y | |
| Extragonadal | II | >11 y | |
| Poor | Testis (IGCCC intermediate or poor risk) | II-IV | >11 y |
| Ovary | IV | >11 y | |
| Extragonadal | III-IV | >11 y | |
| IGCCC = International Germ Cell Consensus Classification. | |||
