Background
Normal enzyme function of N-acetylglutamate synthetase (NAGS) deficiency is confined to the hepatic mitochondria and mediates the reaction acetyl-coenzyme A (CoA) + glutamate → N-acetylglutamate + CoA. As a mitochondrial reaction, each of the substrates is normally omnipresent. Acetyl-CoA is a cofactor in many mitochondrial reactions, and glutamate is the transamination product of α-ketoglutarate and alanine; α-ketoglutarate is produced by the Krebs cycle.
The normal function of N-acetylglutamate (NAG), the reaction product, is to act as an activator of carbamyl phosphate synthetase (CPS), which is also a mitochondrial enzyme. See the image below.
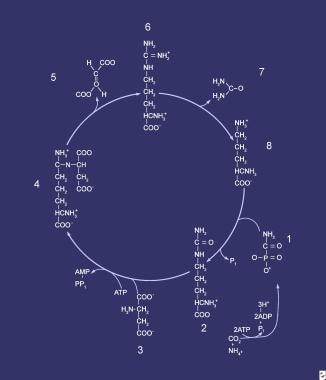 Compounds comprising the urea cycle are numbered sequentially, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
Compounds comprising the urea cycle are numbered sequentially, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
The activation process requires physical binding of NAG to the CPS enzyme, in turn, causing the inactive form of CPS to convert to an active state. Thus, CPS activity is regulated by the relationship of available NAG to inactive CPS enzyme protein.
The biochemical effect of NAGS deficiency is an inability to form adequate NAG; this results in failure to activate the enzyme responsible for the reaction NH4+ + CO2 + ATP → H2 N-CO-PO32- + ADP, which is the entry step into the urea cycle (see Carbamyl Phosphate Synthetase Deficiency). [1]
Clinical signs and symptoms of NAGS deficiency occur when ammonia fails to fix into carbamoyl phosphate (CP) effectively, thus disabling the urea cycle. This leads to accumulation of alanine and glutamine (transamination products of pyruvate and glutamate, respectively) and, finally, of ammonia. The condition is progressive without intervention.
Pathophysiology
Overall, the hepatic urea cycle is the major route for waste nitrogen disposal, generation of which is chiefly from protein and amino acid metabolism. [2] Low-level synthesis of certain cycle intermediates in extrahepatic tissues makes a small contribution to waste nitrogen disposal as well. A portion of the cycle is mitochondrial in nature; mitochondrial dysfunction may impair urea production and result in hyperammonemia. Overall, activity of the cycle is regulated by the rate of synthesis of NAG, the enzyme activator that initiates incorporation of ammonia into the cycle.
Epidemiology
Frequency
United States
Too few cases have been reported to cite any incidence figures. However, recognition of affected patients is increasing. Because the clinical presentation is indistinguishable from that of CPS deficiency and because the diagnosis is difficult, the true incidence may be underestimated. This is further emphasized by the fact that genetically affected individuals may remain asymptomatic for many years.
A newly formed Urea Cycle Disorders Consortium in the United States reported on a cross-section of patients throughout the country; remarkably, not a single case of NAGS deficiency was identified. [3] However, because NAG is the requisite activator for CPS, occasional mistaken diagnoses of CPS deficiency may have obscured cases of NAGS deficiency.
A series of adult cases of NAGS deficiency has been reported, suggesting that some mutations may result in milder clinical variants while complicating accurate diagnosis in children and teens. [4]
International
Only a handful of cases have been reported worldwide.
Mortality/Morbidity
NAGS deficiency is associated with significant morbidity and mortality. Patients who present with hyperammonemia are at risk for cerebral edema and death if treatment is not immediately begun. Survivors of hyperammonemic coma are likely to suffer brain damage and resulting developmental delays, learning disabilities, and/or intellectual disability.
Sex
Case reports of NAGS deficiency have shown the condition to occur in both sexes; because the mutation is inherited as an autosomal recessive trait, this is to be expected.
Age
NAGS deficiency can present at any age. As with many inherited metabolic diseases, the most likely time of presentation is in the newborn period.
Prognosis
Long-term prognosis is unclear; most likely, the future intelligence quotient score depends on the severity of the initial presentation and the subsequent hyperammonemic episodes suffered.
Patient Education
Inform parents of their obligate heterozygote status given the likelihood that this is an autosomal recessive trait.
Parents must understand that the chance of recurrence is 1:4 (25%) with each subsequent pregnancy.
Advise parents to seek early medical attention for the patient in the event of intercurrent illness.
-
Compounds comprising the urea cycle are numbered sequentially, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
