Practice Essentials
Hypoglycemia may be considered a biochemical symptom, indicating the presence of an underlying cause. Because glucose is the fundamental energy currency of the cell, disorders that affect its availability or use can cause hypoglycemia. Hypoglycemia is a common clinical problem in neonates, is less common in infants and toddlers, and is rare in older children. [1, 2] It can be caused by various conditions. The most common cause of mild or severe hypoglycemia in childhood is insulin-treated type 1 diabetes, when there is a mismatch among food, exercise, and insulin. (See Etiology and Epidemiology.)
Complications
Many of the etiologies of hypoglycemia may carry the same consequences, complicating the causal distinction. Infants and children with asymptomatic hypoglycemia have been shown to have neurocognitive defects at the time of hypoglycemia, including impaired auditory and sensory-evoked responses and impaired test performance. (See Prognosis, History, and Physical Examination.)
Long-term consequences of hypoglycemia include decreased head size, lowered IQ, and specific regional brain abnormalities observed using magnetic resonance imaging (MRI).
Physiologic defenses against hypoglycemia
The body normally defends against hypoglycemia by decreasing insulin secretion and increasing glucagon, epinephrine, growth hormone, and cortisol secretion. These hormonal changes combine to increase hepatic glucose output, increase alternative fuel availability, and decrease glucose use (see the diagram below).
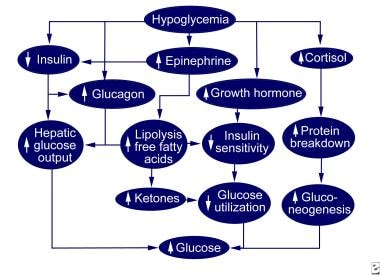
The increase in hepatic glucose production is initially caused by the breakdown of liver glycogen stores resulting from lower insulin levels and increased glucagon levels. When glycogen stores become depleted and protein breakdown increases because of increased cortisol levels, hepatic gluconeogenesis replaces glycogenolysis as the primary source of glucose production. The breakdown of protein is reflected by increased plasma levels of the gluconeogenic amino acids alanine and glutamine. Decreased use of peripheral glucose occurs initially because of a fall in insulin levels and later because of increases in epinephrine, cortisol, and growth hormone levels.
All 3 events increase lipolysis and plasma free fatty acid levels, which are available as an alternative fuel and competitively inhibit glucose use. Increased plasma and urinary ketone levels indicate the use of fat as an energy source. Plasma free fatty acids also stimulate glucose production. Hypoglycemia occurs when 1 or more of these counterregulatory mechanisms fail because of the overuse of glucose (as in hyperinsulinism), the underproduction of glucose (as in the glycogen-storage diseases), or both (as in growth hormone or cortisol deficiency). (See Etiology.)
Signs and symptoms of pediatric hypoglycemia
Signs and symptoms of hypoglycemia in neonates include the following:
-
Tremulousness
-
Brisk Moro reflex
-
Lethargy
-
Poor feeding
-
Irritability
-
Hypothermia
-
Respiratory distress
-
Apnea
-
Bradycardia
-
Seizure
-
Coma
-
Sudden death
Signs and symptoms of hypoglycemia in older children include the following:
-
Dizziness
-
Sweating
-
Hunger
-
Anxiousness
-
Confusion
-
Lethargy
-
Poor feeding
-
Irritability
-
Seizure
-
Coma
-
Sudden death
Diagnosis and management
The ability to properly sort through the differential diagnoses of hypoglycemia depends on obtaining the critical sample at the time of hypoglycemia. This sample is used to measure the various metabolic precursors and hormones involved in glucose counterregulation, including glucose, insulin, growth hormone, cortisol, lactate, pyruvate, beta-hydroxybutyrate, free fatty acid, carnitine, branched-chain amino acid, and insulinlike growth factor-binding protein-1 (IGFBP-1) levels. (A urine sample for organic acid analysis is also critical.)
In hyperinsulinism, positron emission tomography (PET) scanning with [18F] dihydroxyphenylalanine (DOPA) has been shown to effectively distinguish focal from diffuse disease.
In hypopituitarism, head magnetic resonance imaging (MRI) should be performed to identify pituitary or hypothalamic neoplasms or congenital abnormalities.
Short-term treatment of hypoglycemia consists of an intravenous (IV) bolus of dextrose 10% 2.5 mL/kg. The critical sample should be drawn before the glucose is administered. After the bolus is administered, an IV infusion that matches normal hepatic glucose production (approximately 5-8 mg/kg/min in an infant and about 3-5 mg/kg/min in an older child) should be continued. This should be adjusted to maintain the plasma glucose level at more than 3 mmol/L. Children with hyperinsulinemia may have much higher needs. Glucagon infusion at rates of 0.005-0.02 mg/kg/h should be used as a temporary treatment in children with hyperinsulinism in whom adequate amounts of dextrose cannot be given. It can cause a rash and decreased appetite if used over the long term. Long-term care of children with hypoglycemia varies based on the etiology.
For hypoglycemia in patients with diabetes, treatment depends on the patient's mental status. If the patient is awake and alert, 15 g of simple carbohydrate (4 oz of most fruit juices, 3 tsp of sugar, glucose tablets) by mouth should be sufficient. If the patient's mental status is altered and aspiration is a concern, treatment depends on the patient's setting. At home, intramuscularly administered glucagon is the best choice and should be available to families or close associates of all insulin-treated patients with diabetes. In the hospital setting, IV dextrose 25% is appropriate treatment.
Surgery for hyperinsulinism is usually performed when medical therapy fails or when the patient is an older child with a possible insulin-producing tumor.
Dietary prevention of hypoglycemia depends on the underlying condition. In patients with a metabolic disease, avoidance of specific substances is usually necessary and is dependent on the specific condition.
Etiology
Hyperinsulinemia
Hyperinsulinemia is characterized by an excessive use of glucose. Possible causes of hyperinsulinism in children include maternal diabetes in pregnancy, persistent hyperinsulinemic hypoglycemia of infancy, insulin-producing tumors, and child abuse. Hyperinsulinism causes excess glucose use primarily by stimulating skeletal muscle to take up glucose. This is aggravated by insulin-induced suppression of hepatic glycogenolysis and gluconeogenesis. [3]
In infants, hyperinsulinemia may be due to various genetic defects that cause a loss of glucose regulation of insulin secretion. This disorder is known as endogenous-persistent hyperinsulinemic hypoglycemia of infancy (previously termed nesidioblastosis). The most common of these disorders is associated with an inactive or only partially active potassium channel. This channel is composed of 2 parts: the sulfonylurea receptor (SUR1) and the potassium pore (Kir6.2). The former is encoded by the ABCC8 gene, and the latter by the KCNJ11 gene. [4]
No genetic defect is identified in 50% of patients with hyperinsulinism, although unusual single nucleotide polymorphisms defects have been found that may be responsible in some infants. [5]
Infants of mothers with diabetes also have high insulin levels after birth due to the high glucose exposure in utero; the poorer the glucose control during pregnancy, the greater the likelihood of hyperinsulinism in the infant. In older children, hyperinsulinemia is rare, but an insulin-producing tumor is the most common cause. Exogenous administration of insulin or oral hypoglycemic agents, either accidental or due to abuse, must be considered.
Hyperinsulinemia can also result from in utero or perinatal stress (eg, maternal hypertension, prematurity, small for gestational age, hypoxia, Caesarian delivery). [6] These infants have no genetic mutations. Hypoglycemia is temporary and resolves in 1-14 months. The mechanism is unclear.
Uncommon genetic causes of hyperinsulinism in infants
Activating defects of the GCK gene for the enzyme glucokinase, which serves as the primary glucose sensor in the β cell, are rare. Most of these defects are autosomal recessive, but some are autosomal dominant. This defect causes an increased intracellular ATP/adenosine diphosphate (ADP) ratio and closure of the potassium-ATP channel.
Defects in GLUD1, which encodes the enzyme glutamate dehydrogenase, are usually associated with asymptomatic hyperammonemia and cause hyperinsulinism; however, the relationship is not entirely understood. Noninfant children with these gene defects also have asymptomatic hyperammonemia.
Genetic defects in the enzyme short-chain L-3-hydroxyacyl-CoA dehydrogenase and the transcription factor hepatocyte nuclear factor 4 alpha have been described in patients with hypoketotic hyperinsulinemic hypoglycemia, although the causal mechanisms are unknown.
A defect in SLC16A1 (monocarboxylate transporter) may cause exercise-induced hyperinsulinemic hypoglycemia.
Metabolic defects
Glucose-processing defects (Krebs cycle defects, respiratory chain defects) are rare; they interfere with the ability to appropriately generate adenosine triphosphate (ATP) from glucose oxidation. Lactate levels are high.
Defects in alternative fuel production (eg, carnitine acyl transferase deficiency, hepatic hydroxymethyl glutaryl coenzyme A [HMG CoA] lyase deficiency, long-chain and medium-chain acyl-coenzyme A dehydrogenase deficiency, variably in short-chain acyl-coenzyme A dehydrogenase deficiency) interfere with the use of fat as an energy supply, meaning that the body depends only on glucose. This becomes a problem during periods of prolonged fasting that frequently accompany gastrointestinal (GI) illness. These defects are frequently tested for during neonatal screening. Sepsis or other hypermetabolic states, such as hyperthyroidism, may cause hypoglycemia.
Disorders of glucose underproduction
Inadequate glucose stores are associated with prematurity, infants who are small for gestational age, malnutrition, and ketotic hypoglycemia. After insulin treatment in diabetes, these disorders are the most common causes of hypoglycemia. The first three of these should be readily apparent based on the clinical situation. Ketotic hypoglycemia, which usually affects small, thin children aged 18 months to 6 years, is usually caused by disrupted food intake. Ketotic hypoglycemia is a diagnosis of exclusion, made after other causes of hypoglycemia are ruled out.
Glycogen synthase deficiency (glycogen-storage disease type 0) is associated with fasting hypoglycemia because of the liver’s inability to store glucose in the immediate postprandial state. Thus, the glucose load from the meal is anaerobically metabolized rather than stored for later use. In this disorder, plasma glucose and lactate levels are high in the immediate postprandial state. Glycogen synthase deficiency must be considered before the diagnosis of ketotic hypoglycemia is assigned.
Disorders of hepatic glucose production include glucose-6-phosphatase deficiency (glycogen-storage disease type Ia); glucose 6-phosphate translocase deficiency (glycogen storage disease type 1b); debrancher deficiency (glycogen-storage disease type III); hepatic phosphorylase deficiency (glycogen-storage disease type VI); glycogen synthase deficiency; fructose 1,6 diphosphatase deficiency; phosphoenolpyruvate deficiency; pyruvate carboxylase deficiency; galactosemia; hereditary fructose intolerance; and maple syrup urine disease. These disorders interfere in glucose production through various defects, including blockage of glucose release or synthesis or blockage or inhibition of gluconeogenesis. Children with these diseases may adapt to their hypoglycemia because of its chronicity.
Hormonal abnormalities include panhypopituitarism, growth hormone deficiency, and cortisol deficiency (primary or secondary). As described above, growth hormone and cortisol play important roles in generating alternative fuels and stimulating glucose production. Because they are easily treatable abnormalities, early recognition is important.
Toxins, corticosteroids, and other illnesses
Hypoglycemia can also be caused by toxins (including ethanol, salicylates, propranolol) and illnesses (eg, malaria). Ethanol inhibits gluconeogenesis in the liver and can thus cause hypoglycemia. (This is particularly true in patients with insulin-treated diabetes who are unable to reduce insulin secretion in response to developing hypoglycemia.) Salicylate intoxication causes hyperglycemia and hypoglycemia. The latter is due to augmentation of insulin secretion and inhibition of gluconeogenesis.
A retrospective study by Zigron et al found that among preterm infants exposed to antenatal corticosteroid, 23.2% were found to have neonatal hypoglycemia. Multivariate analysis indicated that delivery within 24-48 hours after corticosteroid administration is an independent risk factor for hypoglycemia in neonates, with 40% of the study’s newborns delivered within this time interval being hypoglycemic, versus 22% of those delivered outside the interval. [7, 8]
Large for gestational age in mother with type 1 diabetes
A retrospective cohort study by Yamamoto et al of 161 women with type 1 diabetes mellitus indicated that in women with type 1 diabetes, large-for-gestational-age infants have a 2.5-fold greater risk for neonatal hypoglycemia. The study suggested that being large for gestational age in neonates of mothers with type 1 diabetes may be better than maternal glycemic control in terms of predicting neonatal hypoglycemia. [9]
Epidemiology
One study found hypoglycemic events in newborns weighing more than 2500 grams from term, singleton, nondiabetic pregnancies occurring at rate of 24.7 events per 1000 infant-days at risk. [10] Hypoglycemia is more common in neonates born at less than 37 weeks' gestation and in those born at more than 40 weeks' gestation, with incidence rates of 2.4% in neonates born at 37 weeks' gestation, 0.7% in neonates born at 38-40 weeks' gestation, and 1.6% and 1.8% in neonates born at 41 weeks' gestation and 42 weeks' gestation, respectively. [11] The incidence of hypoglycemia in children older than 6 months in a large urban emergency department was 0.034%. [12]
A study by Birkebaek et al found the rate of severe hypoglycemia among children with type 1 diabetes mellitus in Denmark, Iceland, Norway, and Sweden to be 6.0 per 100 patient-years, with the lowest incidence in Sweden. [13]
Age-related demographics
Hypoglycemia is most common in the immediate postneonatal period. The incidence of new cases decreases with increasing age, and true hypoglycemia is extremely rare in adolescents. Patient age is also helpful in assessing the probable diagnosis of hypoglycemia. Hyperinsulinemia, hypopituitarism, and inborn errors of metabolism are frequent causes of hypoglycemia in infancy. In toddlers, ketotic hypoglycemia is most common. In adolescents, insulin-producing pancreatic tumors are the most common cause of true hypoglycemia.
Prognosis
Prognosis clearly is dependent on the underlying condition. Inborn errors of metabolism and hormonal deficiencies are lifelong diseases that require lifelong treatment. On the other hand, ketotic hypoglycemia is generally outgrown when the child has adequate nutritional stores to prevent hypoglycemia, which is usually around age 5 years.
The prognosis in hyperinsulinism varies and depends on the severity of the disease, whether it is amenable to medical therapy, and whether the lesion is focal or diffuse. Focal lesions can frequently be surgically cured. Mild hyperinsulinism that is responsive to diazoxide may require long-term therapy but may allow the child to lead a normal life. Diffuse lesions that are not responsive to medical therapy are frequently not entirely cured by pancreatectomy and may present continued problems, including hypoglycemia and developmental delay or, at the opposite extreme, type 1 diabetes.
In a study of patients under age 20 years with type 1 diabetes, Pacaud et al found that in those who suffered an episode of severe hypoglycemia/hypoglycemic coma, the risk for another such episode remained higher than that in patients who never had an episode even 4 years after the episode occurred. [14]
Morbidity
Hypoglycemia has both acute and long-term consequences (see Clinical). Infants and children with asymptomatic hypoglycemia have been shown to have neurocognitive defects at the time of hypoglycemia, including impaired auditory and sensory-evoked responses and impaired test performance. Many etiologies of hypoglycemia may have the same consequences, complicating the causal distinction.
Long-term consequences of hypoglycemia include decreased head size, lowered intelligence quotient (IQ), and specific regional brain abnormalities revealed by MRI. As many as 50% of patients who survive hyperinsulinemic hypoglycemia of infancy have long-term neurologic complications; this rate has changed little since the end of the 20th century. This emphasizes the need for early recognition and treatment of these children.
A study by Mahajan et al indicated that mean motor development and mental development quotients at corrected ages 6 and 12 months tend to be lower in neonates with symptomatic or asymptomatic hypoglycemia than in neonates with euglycemia. Moreover, these quotients were found to be lower in symptomatic infants than in asymptomatic ones. [15]
However, a study by Goode et al indicated that neurodevelopmental outcomes in preterm infants with neonatal hypoglycemia do not significantly differ from those of preterm infants who are euglycemic. The study, which compared preterm infants with hypoglycemia with controls at ages 3, 8, and 18 years, found no significant differences in cognitive or academic skills at any age. In addition, no clinically meaningful difference in problem behaviors was found. [16]
-
Normal hypoglycemic counterregulation.
-
Interpretation of the critical sample.
Tables

What would you like to print?
- What Are the Priorities of People With Type 1 Diabetes?
- Are Immune Therapies for Type 1 Diabetes Worthwhile?
- 'No Excuses': Training for the Olympics with Type 1 Diabetes
-
 Choosing Which Agents to Use in the Management of CKD in Type 2 Diabetes Patients: SGLT2 Inhibitors vs GLP-1s
Choosing Which Agents to Use in the Management of CKD in Type 2 Diabetes Patients: SGLT2 Inhibitors vs GLP-1s
- Anticoagulation and Antiplatelet Therapy for Atrial Fibrillation and Stable Coronary Disease
-
Apr 04, 2025 This Week in Cardiology Podcast


 Complications of Diabetes
Complications of Diabetes


