Practice Essentials
Hypogonadism refers to a condition in which little or no hormone is produced by the testes or ovaries. The condition can be hypergonadotropic (primary, resulting when the gonads fail) or hypogonadotropic. The latter can result from failure of the hypothalamic luteinizing-hormone releasing hormone [LHRH] pulse generator or from the inability of the pituitary to respond with secretion of luteinizing hormone [LH] and follicle-stimulating hormone [FSH]. (See the image below.) Morbidity for men and women with hypogonadism includes infertility and an increased risk of osteoporosis; there is no increase in mortality.
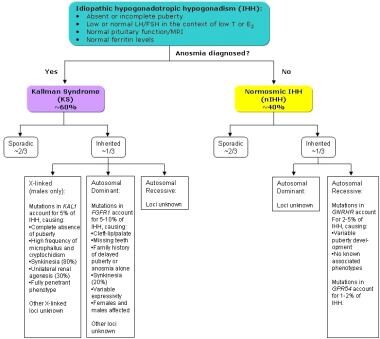 Types of idiopathic hypogonadotropic hypogonadism.
Types of idiopathic hypogonadotropic hypogonadism.
Causes of hypogonadism
Hypogonadotropic hypogonadism can result from the following:
-
Central nervous system (CNS) disorders - Including tumors and miscellaneous causes involving the pituitary/hypothalamic area
-
Genetic causes - Eg, Kallmann syndrome
-
Congenital disorders - Eg, Prader-Willi syndrome, Laurence-Moon syndrome, Bardet-Biedl syndrome, [1] and Gaucher disease
-
Acquired disorders - Eg, exercise-induced hypogonadism, psychogenic hypogonadism, hyperprolactinemia, Cushing syndrome, human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS), morbid obesity, type II diabetes mellitus
Causes of hypergonadotropic hypogonadism (primary hypogonadism) in males include the following:
-
Klinefelter syndrome
-
Inactivating mutation
-
Chemotherapy
-
Radiation therapy
-
Gonadectomy
-
Anorchism and cryptorchidism
-
Testicular biosynthetic defects - 17β-hydroxylase dehydrogenase deficiency, 5α-reductase deficiency, 17-hydroxylase deficiency
-
Defects in testicular determination - Gonadal dysgenesis
-
Other rare disorders of sex development (DSDs) - Ovotesticular DSD, XX males
-
Sertoli-cell-only syndrome
-
LH resistance
Causes of hypergonadotropic hypogonadism (primary hypogonadism) in females include the following:
-
Turner syndrome
-
Inactivating mutations
-
XX and XY gonadal dysgenesis
-
Premature menopause
-
Radiation therapy
-
Chemotherapy
-
Autoimmune oophoritis
-
Resistant ovary
-
Galactosemia
-
Glycoprotein syndrome type 1
-
FSH-receptor gene mutations
-
LH/human chorionic gonadotropin (hCG) resistance
-
Polycystic ovarian disease
Signs and symptoms of hypogonadism
History
Considerations in the evaluation of males with hypogonadism include the following:
-
Developmental anomalies associated with the genital system (eg, hypospadias, micropenis, and cryptorchidism) [2]
-
For postpubertal males, the rate of beard growth, libido and sexual function, muscle strength, and energy levels
-
Possible causes of acquired testicular failure (eg, mumps orchitis, trauma, radiation exposure of the head or testes, and chemotherapy)
-
Drugs that may interrupt testicular function - Including agents that interfere with testosterone synthesis, such as spironolactone and cyproterone. Agents such as cortisol, marijuana, heroin, and methadone may interfere with gonadotropin secretion.
Considerations in the evaluation of females with hypogonadism include the following:
-
Signs associated with Turner syndrome (eg, lymphedema, cardiac or renal congenital anomalies, and short growth pattern)
-
Age of menarche
Physical examination
Considerations in the physical examination of males with hypogonadism include the following:
-
Evaluation of the testes: This is the most important feature of the physical examination; determine whether both testes are palpable, their position in the scrotum, and their consistency; testes size can be quantitated by comparison with testicular models (orchidometer), or their length and width may be measured
-
Examination of the genitalia for hypospadias
-
Examination of the scrotum to see if it is completely fused
-
Evaluation of the extent of virilization
-
Staging of puberty: Use the Tanner criteria for genitalia, pubic hair, and axillary hair
-
Examination for signs of Klinefelter syndrome (eg, tall stature, especially if the legs are disproportionately long, gynecomastia, small or soft testes, and a eunuchoid body habitus)
Considerations in the physical examination of females with hypogonadism include the following:
-
Examination of the genitalia is important
-
Determination of the extent of androgenization: May be adrenal or ovarian in origin and is demonstrated in pubic and axillary hair
-
Determination of the extent of estrogenization: As evidenced by breast development and maturation of the vaginal mucosa
-
Examination for signs of Turner syndrome (eg, short stature, webbing of the neck [such as pterygium colli], a highly arched palate, short fourth metacarpals, widely spaced nipples, or multiple pigmented nevi)
See Clinical Presentation for more detail.
Diagnosis of hypogonadism
The following studies may be indicated in males with hypogonadism:
-
Follicle-stimulating hormone (FSH) levels
-
Luteinizing hormone (LH) levels
-
Prolactin levels
-
Testosterone levels
-
Thyroid function
-
Seminal fluid examination
-
Karyotyping
-
Testicular biopsy
For males after puberty, the Guidelines of the Endocrine Society [3] require that the diagnosis of hypogonadism be based on symptoms and signs of hypogonadism plus the presence of a low testosterone level measured on at least 2 occasions.
The following studies may be indicated in females with hypogonadism:
-
FSH levels
-
LH levels
-
Estradiol levels
-
Antiovarian antibody levels: If gonadotropin levels are elevated
-
Thyroid function
-
Karyotyping
Additional tests in the evaluation of patients with hypogonadism include the following:
-
Adrenocorticotropic hormone (ACTH) stimulation testing: In patients in whom a form of congenital adrenal hyperplasia is suspected, adrenal steroid synthesis is best evaluated by performing a cosyntropin (ACTH 1-24) stimulation test
-
Luteinizing-hormone releasing hormone (LHRH) stimulation testing: To distinguish between true hypogonadotropic hypogonadism and constitutional delay in growth and maturation
-
Testicular tissue testing: If the testes are not palpable and if it is not certain whether any testicular tissue is present, administering human chorionic gonadotropin (hCG) and measuring testosterone response may be helpful
See Workup for more detail.
Management
Hormonal replacement
The simplest and most successful treatment for males and females with either hypergonadotropic or hypogonadotropic hypogonadism is replacement of sex steroids, but the therapy does not confer fertility or, in men, stimulate testicular growth.
When fertility is desired, an alternative therapy for men with hypogonadotropic hypogonadism is administration of pulsatile LHRH or injections of hCG and FSH. (In patients with hypergonadotropic hypogonadism [primary hypogonadism], fertility is not possible.)
In a 6-year European study of men being treated for hypogonadism, long-term transdermal testosterone treatment did not increase prostate-specific antigen (PSA) levels or influence prostate cancer risk. [4, 5]
Investigators used data from a 5-year, open-label extension of a 1-year trial of a transdermal testosterone patch (Testopatch) in men with hypogonadism. Study subjects wore two 60 cm2 patches, each of which delivered 2.4 mg of testosterone per day. More than 90% of patients had PSA concentrations below 2 ng/mL during the 6-year study, and no prostate cancer was found in patients over the course of the trial.
See Treatment and Medication for more detail.
Background
Hypogonadism manifests differently in males and in females before and after the onset of puberty. [6] If onset is in prepubertal males and testosterone replacement is not instituted, the individual has features of eunuchoidism, which include sparse body hair, poor development of skeletal muscles, and delay in epiphyseal closure, resulting in long arms and legs. When hypogonadism occurs in postpubertal males, lack of energy and decreased sexual function are the usual concerns. In females with hypogonadism before puberty, failure to progress through puberty or primary amenorrhea is the most common presenting feature. When hypogonadism occurs in postpubertal females, secondary amenorrhea is the usual concern.
Pathophysiology
The gonads (ovaries or testes) function as part of the hypothalamic-pituitary-gonadal axis. A hypothalamic pulse generator resides in the arcuate nucleus, which releases luteinizing hormone (LH)-releasing hormone (LHRH), which is also termed gonadotropin-releasing hormone (GnRH), into the hypothalamic-pituitary portal system. Data suggest that a gene named KISS is important in the development of the LHRH-secreting cells. [7, 8]
In response to these pulses of LHRH, the anterior pituitary secretes follicle-stimulating hormone (FSH) and LH, which, in turn, stimulate gonadal activity. The increase in gonadal hormones results in lowered FSH and LH secretion at the pituitary level, completing the feedback loop. In the testes, LH stimulates Leydig cells to secrete testosterone, whereas FSH is necessary for tubular growth. In the ovaries, LH acts on theca and interstitial cells to produce progestins and androgens, and FSH acts on granulosa cells to stimulate aromatization of these precursor steroids to estrogen.
Hypogonadism may occur if the hypothalamic-pituitary-gonadal axis is interrupted at any level. Hypergonadotropic hypogonadism (primary hypogonadism) results if the gonad does not produce the amount of sex steroid sufficient to suppress secretion of LH and FSH at normal levels. Hypogonadotropic hypogonadism may result from failure of the hypothalamic LHRH pulse generator or from inability of the pituitary to respond with secretion of LH and FSH. Hypogonadotropic hypogonadism is most commonly observed as one aspect of multiple pituitary hormone deficiencies resulting from malformations (eg, septooptic dysplasia, other midline defects) or lesions of the pituitary that are acquired postnatally. In 1944, Kallmann and colleagues first described familial isolated gonadotropin deficiency. Recently, many other genetic causes for hypogonadotropic hypogonadism have been identified.
Normosmic hypogonadotropic hypogonadism, in which the sense of smell is not disrupted, has been associated with mutations in GNRH1, KISS1R, and GNRHR genes. Although their exact functions are unclear, the genes TAC3 and TACR3 have also been associated with normosmic hypogonadotropic hypogonadism. Kallmann syndrome (anosmic hypogonadotropic hypogonadism) has been associated with mutations in KAL1, FGFR1, FGF8, PROK2, and PROKR2 genes. The relationship with Kallmann syndrome is thought to be because these genes are all related to the development and migration of GnRH neurons. Mutations of an additional gene, CHD7, which has been associated with CHARGE syndrome, has also been found in patients with normosmic or anosmic hypogonadotropic hypogonadism.
Frequency
In women with hypergonadotropic hypogonadism (ie, gonadal failure), the most common cause of hypogonadism is Turner syndrome, which has an incidence of 1 case per 2,500-10,000 live births. In men with hypergonadotropic hypogonadism, the most common cause is Klinefelter syndrome, which has an incidence of 1 case per 500-1000 live births. Hypogonadotropic hypogonadism is rarer.
A study by Livingston et al found that among men whose testosterone levels were checked in primary health-care exams, potential hypogonadism was found in a significant minority. The investigators reported a total testosterone level of under 10 nmol/L in 1924 out of 8788 men (21.9%) in whom testosterone results were determined. [9]
Epidemiology
No racial predilection has been described.
Hypergonadotropic hypogonadism is more common in males than in females because the incidence of Klinefelter syndrome (the most common cause of primary hypogonadism in males) is higher than the incidence of Turner syndrome (the most common cause of hypogonadism in females). Incidence of hypogonadotropic hypogonadism is equal in males and females.
Hypogonadism may occur at any age; however, consequences differ according to the age at onset. If hypogonadism occurs prenatally (even if incomplete), sexual ambiguity may result. If hypogonadism occurs before puberty, puberty does not progress. If hypogonadism occurs after puberty, infertility and sexual dysfunction result.
Prognosis
No increase in mortality is observed in patients with hypogonadism. Morbidity for men and women includes infertility and an increased risk of osteoporosis. In women, an increased risk of severe osteoporosis is noted. In men, hypogonadism causes decreased muscle strength and sexual dysfunction.
Men and women with hypogonadism can lead a normal life with hormone replacement.
Approximately 10-20% of females with Turner syndrome have some spontaneous puberty. Spontaneous estrogenization occurs more commonly in women with mosaic karyotypes and those karyotypes with an abnormal second X chromosome, such as 46,XXiq or 46,XXip. Reports exist of women with mosaic Turner syndrome becoming pregnant without in vitro fertilization.
A multicenter, international, cross-sectional study by Dwyer et al indicated that in males with congenital hypogonadotropic hypogonadism, certain predictors suggest whether or not a patient will experience spontaneous reversal of the hypogonadism after treatment. The investigators found it more likely that anosmia, cryptorchidism, complete absence of puberty (testicular volume < 4 cm3), and at least two rare genetic variants (ie, oligogenicity) would exist in males who do not undergo reversal. In those study patients who were genetically tested, none who experienced reversal had a rare pathogenic form of the ANOS1 gene, compared with 10.5% of those who did not undergo reversal, while those in the reversal group did demonstrate a significantly greater rate of rare variants in the GNRHR gene (12.0% of tested patients) than did the non-reversal patients (3.2% of tested patients). [10]
Patient Education
For patient education resources, see the Men's Health Center and Women's Health Center, as well as Impotence/Erectile Dysfunction and Amenorrhea.
-
Types of idiopathic hypogonadotropic hypogonadism.
