Practice Essentials
Menkes disease, also known as kinky hair disease, is an X-linked neurodegenerative disease of impaired copper transport.
Signs and symptoms
Children with the classic form of Menkes disease usually present at 2–3 months of age with the following:
-
Loss of developmental milestones
-
Profound truncal hypotonia
-
Failure to thrive
Findings include abnormal kinky hair, eyebrows, and eyelashes (see the image below).
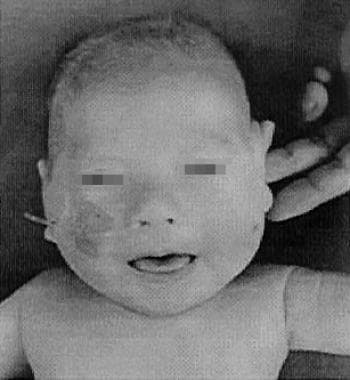 Four-month-old patient with classic Menkes disease. His hair is depigmented and lusterless with pili torti and the skin is pale with eczema.
Four-month-old patient with classic Menkes disease. His hair is depigmented and lusterless with pili torti and the skin is pale with eczema.
Diagnosis
Diagnostic findings are as follows:
-
Serum copper level less than 70 mg/dL (reference 80–160)
-
Serum ceruloplasmin level less than 20 mg/dL (reference 20–60)
Management
There is no cure for Menkes disease, but treatment with copper supplements can help manage symptoms and increase survival.
Treatment with copper chloride and/or L-histidine should be provided by a clinician familiar with their use. Copper chloride and L-histidine solutions of 350–500 µg/d or qod injected intravenously or subcutaneously increase the serum and cerebrospinal fluid copper levels to the normal range after 6 weeks. Patients with a genetic abnormality that are still able to produce limited amounts of ATP7A may receive benefit from early treatment.
Background
Menkes disease, also known as kinky hair disease, is an X-linked neurodegenerative disease of impaired copper transport. Menkes et al first described it in 1962. Danks et al first noted that copper metabolism is abnormal in 1972; in 1973, after noting the similarity of kinky hair to the brittle wool of Australian sheep raised in areas with copper-deficient soil, he demonstrated abnormal levels of copper and ceruloplasmin in these patients.
A girl with the Menkes disease phenotype and an X:autosome chromosomal translocation was described in 1987, which led to the identification of the locus on the X chromosome in 1993. Milder variants of Menkes disease, including occipital horn syndrome (also known as X-linked cutis laxa or Ehlers-Danlos type 9) have also been described. The brindled mouse, viable brindled mouse, and blotchy mouse are animal models of the classic form, the mild form, and the occipital horn syndrome, respectively.
The image below depicts an infant with Menkes disease.
Four-month-old patient with classic Menkes disease. His hair is depigmented and lusterless with pili torti and the skin is pale with eczema.
See 21 Hidden Clues to Diagnosing Nutritional Deficiencies, a Critical Images slideshow, to help identify clues to conditions associated with malnutrition.
Pathophysiology
Copper is a trace metal in many essential enzyme systems, including cytochrome C oxidase, superoxide dismutase, lysyl oxidase, tyrosinase, ascorbic acid oxidase, ceruloplasmin, and dopamine beta hydroxylase. The deficiency or impaired function of these enzyme systems is thought to be responsible for the clinical findings of Menkes disease. The Menkes gene is located on the long arm of the X chromosome at Xq21.1, and the gene product (ATP7A) is a 1500–amino acid P-type adenosine triphosphatase (ATPase) that has 17 domains – 6 copper binding, 8 transmembrane, a phosphatase, a phosphorylation, and an ATP binding.
The Menkes and Wilson disease genes have 55% amino acid identity. Menkes and Wilson disease ATPases use common biochemical mechanisms, but the tissue-specific expression differs. The Wilson disease gene (WND) is expressed predominantly in the liver, whereas the Menkes disease gene (MNK) is not expressed in the liver. The predominant sites of Menkes gene expression are the placenta, GI tract, and blood-brain barrier. The Menkes protein is also in retinal pigment epithelial cells and the neurosensory retina. [1]
Hardman et al found that insulin and estrogen increased the level of MNK mRNA and protein levels in the placenta. [2] The MNK protein also became localized toward the basolateral membrane and increased copper transport. Hardman et al found levels of the Wilson disease ATPase decreased in response to insulin and was perinuclear. His conclusion was that the MNK protein delivers copper to the fetus and Wilson disease ATPase returns excess copper to the maternal circulation.
Niciu et al found that ATP7A is most abundant in the early postnatal period and peaks at P4 in the neocortex and cerebellum in the mouse brain. [3] ATP7A levels are highest in the ependymal cells of the lateral and third ventricles. ATP7A increases in CA2 hippocampal pyramidal and cerebellar Purkinje neurons but decreases in other cell populations postnatally. Schlief et al found that copper is protective and copper chelation exacerbates NMDA-mediated excitotoxic cell death in hippocampal neurons. [4]
All copper-transporting ATPases have a histidine residue in the large cytoplasmic loop adjacent to the ATP-binding domain. The histidine residue is the most common mutation site in Wilson disease, and this histidine residue is essential for the function of the Menkes ATPase, ATP7A. The Menkes protein is synthesized as a single-chain polypeptide localized to the trans-Golgi network in cells.
Under normal circumstances, ATP7A transports copper into the secretory pathway of the cell for incorporation into the cuproenzymes and excretion from the cell. An increase in intracellular copper causes ATP7A to move to the plasma membrane. As the copper is concentrated into vesicles for excretion from the cell, the cytosolic copper concentration decreases and ATP7A returns to the trans-Golgi network. The migration of ATP7A appears to involve amino acid sequences in the carboxyl terminus, utilizing both clathrin-dependent and clathrin-independent endocytosis. Menkes disease can be caused by both copper transport dysfunction and abnormal protein trafficking.
In Menkes disease, transport of dietary copper from intestinal cells is impaired, leading to the low serum copper levels. Abnormal copper transport in other cells leads to paradoxical copper accumulation in duodenal cells, kidney, pancreas, skeletal muscle, and placenta.
The Menkes gene product protein exists in both a truncated and a long form. The truncated form, which is located in the endoplasmic reticulum, is present in the occipital horn syndrome. The partial preservation of the copper transport-ATPase activity may account for the milder phenotype. Exon skipping is common in Menkes disease. Normal splicing of mRNA depends on a highly conserved, 9-nucleotide splice donor sequence. Slight variations from the splice donor sequence are common, except for the invariant GT (guanine-thiamine) dinucleotide at the +1 and +2 intronic positions. Splice acceptor sites have an invariant AG (adenine-guanine) dinucleotide at intronic positions -1 and -2. Splice junction mutations of the invariant bases severely reduce correct splicing. Patients with the milder Menkes phenotypes have mutations at other sites so that proper splicing of some protein still occurs.
Gross gene deletions occur in about 15% of patients, usually with the classic form of Menkes disease. The point mutations in ATP7A are 23% at the splice site, 20.7% nonsense, 17.2% missense, and 39.1% small deletions/insertions. Variable expression of an identical mutation can be present within a family.
Deficient cytochrome C oxidase (CCO) activity probably accounts for most of the neurologic symptoms, similar to patients with Leigh disease (subacute necrotizing encephalomyelopathy) who have reduced or absent CCO activity and similar neuropathologic changes. Linnebank et al found that copper supplementation in vitro helped to decrease homocysteine toxicity and preserved CCO activity. [5] Decreased lysyl oxidase (LO) activity accounts for the connective-tissue fragility and vascular abnormalities in Menkes disease, since LO deaminates lysine and hydroxylysine in the first step for collagen cross-linkage. LO localizes to the trans-Golgi network so subcutaneous injections of copper-histidine do not improve the activity of LO as the copper is not delivered into the Golgi apparatus. Tyrosinase deficiency (involved in melanin biosynthesis) most likely accounts for the hypopigmentation of the hair and skin seen in Menkes disease.
Purkinje cell loss is profound in Menkes disease. During development, the expression of ATP7A switches from Purkinje cells to Bergmann glia cells, which support Purkinje cell function in adults. ATP7A is a faster copper transporter and catalyses reactions faster than ATP7B, the deficient protein in Wilson disease. Peptidylglycine alpha-amidating mono-oxygenase (PAM) is necessary for the removal of the glycine residue from neuroendocrine precursors—corticotropin-releasing factor (CRH), thyrotropin-releasing hormone (TRH), calcitonin, and vasopressin. Deficiency of dopamine-beta-hydroxylase leads to reduced catecholamine levels. Decreased ascorbic acid oxidase activity leads to bone changes similar to those seen in scurvy.
Moller et al found that even a low level (2-5%) of normally spliced Menkes protein was sufficient to produce the milder occipital horn syndrome in one patient. [6] In contrast, the classic phenotype was seen in 2 patients with a similar mutation who did not produce any normal Menkes protein. Tang et al found that a novel occipital horn syndrome mutation (N1304S) was associated with approximately 30% residual function of ATP7A. [7]
Etiology
Menkes disease is caused by mutations of the Xq21.1 gene (ATP7A).
Many of the gene defects are deletions that can be detected by Southern blot, but small duplications, nonsense mutations, and missense mutations have also been reported.
The mild variants of the disease in humans are generally caused by splicing defects.
Epidemiology
In the United States, incidence is 1 in 50,000 to 1 in 250,000; one third of cases result from new mutations. A study in Japan from 1993 to 2003 found that Menkes disease incidence was 1 per 2.8 million live births and 4.9 per million male live births. [8]
Menkes disease is an X-linked recessive condition and, therefore, usually affects boys through unaffected carrier women. The disease can be due to germ line mosaicism.
A few affected females with X: autosome translocations, X0/XX mosaicism, or unfavorable lyonization have been reported.
The onset of the classic form is in infancy. The milder variants have their onset in childhood or early adulthood.
Prognosis
Most untreated patients with the classic form of Menkes disease die by age 3 years.
Kaler et al noted that infants who received treatment early (within 10 +/- 4 days) had a 92% survival after a median follow-up of 4.6 years. Historical controls that were diagnosed at 163 +/- 113 days and treated only had a 13% survival after a median follow-up of 1.8 years. In fact, 2 of their patients (one with a missense mutation and one with a splice junction alteration) who were treated within 10 days of life had normal neurodevelopmental outcomes at age 5 and 7. A child with the same mutation as the 5-year-old but treated later at 22 days of age did have neurologic sequelae but could walk with support and ride a tricycle. Patients with mutations that disrupt the translational reading frame or have a premature termination codon did not do as well. Their treatment regimen consisted of subcutaneous copper histidine injections of 250 micrograms twice a day to 1 year of age and then daily injections to 3 years of age. [9]
Tang et al also reported that 2 infants treated within 25 days of age had near normal neurodevelopment at 7 months and 3 years of age. However, another infant with an identical missense mutation who began treatment at 228 days of age remained at a 2- to 4-month neurodevelopmental level at a chronological age of 2.5 years. This mutation resulted in normal levels of ATP7A transcript, but the mutated protein had abnormally high posttranslational degradation. [7]
Paulsen et al caution that the biochemical profile needs to be examined as reinitiation of protein translation may ameliorate the effects of a large frameshift deletion with a premature termination codon. [10]
Donsante et al also reported that 3 untreated brothers had differing courses of their disease. The 2 older brothers had mild disease and were able to ambulate independently and did not have seizures. The youngest brother was more delayed and had epilepsy, but he also had a cardiac arrest as a neonate. The cardiac arrest may be the cause of the more severe problems in the youngest brother, but he also had lower serum copper levels and the highest DOPA:DHPG ratio. Furthermore, Donsante et al also reported 2 untreated brothers who had different courses. The older brother also had a milder course with the occipital horn phenotype but the younger brother had profound delay and epilepsy. The less affected sibling had higher ATP7A levels but it was not clear why. There was no somatic mosaicism detected in either family. [11]
In 2017, Tumer et at reported on a 37-year-old who was identified as a neonate due to a postive family history. He had a missense variant that caused impaired protein trafficking. He started treatment at 7 weeks of age. He walked at 18 months and started speaking at 38 months. He has had mild gait ataxia since 2 years of age and dysarthria. He is independent in his ADLs. He worked in a sheltered workplace and lived in a supervised community for many years. At the time of the report, he had become in charge of his work section and was living independently. [12]
-
Four-month-old patient with classic Menkes disease. His hair is depigmented and lusterless with pili torti and the skin is pale with eczema.
-
Diverticula of the bladder in a boy with Menkes disease.
-
The clavicles are short with hammer-shaped distal ends in a patient with Menkes disease.
-
Flared metaphyses of the ulna and radius in a 5-month-old patient with classic Menkes disease.
-
Lax skin in a patient with occipital horn syndrome.
-
Occipital horns (arrow) in a 14-year-old boy with occipital horn syndrome.
-
Magnetic resonance imaging of the brain of a patient with Menkes disease. Subdural effusion is evident in the left frontal lesion. Brain atrophy is also evident.
Tables

What would you like to print?
- Optimal Management of Perianal Disease in Crohn's Disease: The Importance of a Multidisciplinary Approach
- Alzheimer’s Disease: The Cholinergic Revival
- Celiac Disease Linked to Persistent Risk for Liver Conditions
-
 Hair Loss: Common Causes and Treatment
Hair Loss: Common Causes and Treatment
- Case #16: Two Sides of the Same Coin
- Scalp Hair Characteristics in the Newborn Infant

