Practice Essentials
Myelodysplastic syndrome (MDS) refers to a heterogeneous group of closely related clonal hematopoietic disorders commonly found in the aging population. All are characterized by one or more peripheral blood cytopenias. Bone marrow is usually hypercellular, but rarely, a hypocellular marrow mimicking aplastic anemia may be seen. Bone marrow cells display aberrant morphology and maturation (dysmyelopoiesis), resulting in ineffective blood cell production.
MDS affects hematopoiesis at the stem cell level, as indicated by cytogenetic abnormalities, molecular mutations, and morphologic and physiologic abnormalities in maturation and differentiation of one or more of the hematopoietic cell lines. [1, 2, 3] See the image below.
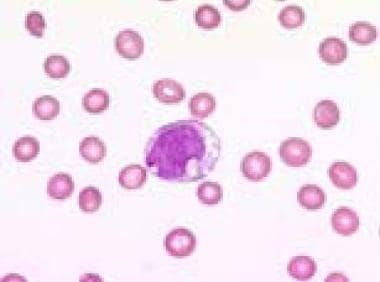 Blood film (1000× magnification) demonstrating a vacuolated blast in a refractory anemia with excess of blasts in transformation. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Blood film (1000× magnification) demonstrating a vacuolated blast in a refractory anemia with excess of blasts in transformation. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
See Myelodysplastic Syndromes: Classification, Features, Diagnosis, and Treatment Options, a Critical Images slideshow, to help identify, classify, work up, and treat these disorders.
MDS may involve one, two, or all three myeloid hematopoiesis cell lineages—erythrocytic, granulocytic, megakaryocytic—depending on the subtype and stage of the disease. The heterogeneity of MDS reflects the fact that its course involves a series of cytogenetic events. In a subgroup of patients, the acquisition of additional genetic abnormalities results in the transformation of MDS into acute myeloid leukemia (AML). Thus, although MDS is clonal, it is considered a premalignant condition.
Patients with MDS may present with clinical manifestations of anemia, thrombocytopenia, and/or neutropenia (see Presentation). The workup in patients with possible MDS includes a complete blood count with differential, peripheral blood smear, and bone marrow studies (see Workup).
Standard care for MDS is constantly changing, but it typically includes supportive therapy, including transfusions, and may include bone marrow stimulation and cytotoxic chemotherapy or hypomethylating agents. Bone marrow transplantation has a limited role. (See Treatment.)
For discussion of MDS in children, see Pediatric Myelodysplastic Syndrome.
For patient education information, see Myelodysplastic or Myeloproliferative Neoplasms (Leukemia Types).
Pathophysiology
MDS develops when a clonal mutation predominates in the bone marrow, suppressing healthy stem cells. The clonal mutation may result from genetic predisposition or from hematopoietic stem cell injury caused by exposure to any of the following:
-
Cytotoxic chemotherapy
-
Radiation
-
Viral infection
-
Genotoxic chemicals (eg, benzene)
MDS can be classified as primary (de novo) or secondary to aggressive treatment of other cancers, with exposure to radiation, alkylating agents, or topoisomerase II inhibitors; it also occurs in heavily pretreated patients with autologous bone marrow transplants.
In the early stages of MDS, the main cause of cytopenias is increased apoptosis (programmed cell death). As the disease progresses and converts into leukemia, further gene mutation occurs, and a proliferation of leukemic cells overwhelms the healthy marrow.
Etiology
Cytogenetically, patients with MDS or AML fall into three groups:
-
Normal karyotype
-
Balanced chromosomal abnormality causing the generation of fusion oncogenes
-
Complex karyotypes (usually >3 abnormalities)
Patients with complex karyotypes constitute 30% of primary MDS cases (only 20% of de novo AML) and up to 50% of therapy-related MDS and AML cases. These patients have a worse prognosis and response to treatment.
Balanced translocation abnormalities lead to the generation of fusion oncogenes such as Bcr-Abl in chronic myelogenous leukemia (CML) and PML-Rar alpha in acute promyelocytic leukemia (APL). Unbalanced recurrent aberrations, most commonly -5, 5q-,-7, 7q-, +8, 11q-, 13q-, and 20q-, suggest that genes within these regions have a role in the pathogenesis of MDS or myeloproliferative disorder (MPD), which is based on loss of tumor suppressor genes or haploinsufficiency of genes necessary for normal myelopoiesis.
Approximately 80% of patients with MDS do not have an obvious exposure or cause for MDS. In these cases, the disorder is classified as primary or idiopathic MDS.
The World Health Organiztion (WHO) classifies secondary MDS as MDS or acute leukemia that develops years after known exposure to sources of chromosomal damage. Patients who survive cancer treatment with alkylating agents, with or without radiotherapy, have a high risk of developing MDS or secondary acute leukemia 5-7 years after the exposure. These drugs are associated with a high prevalence of chromosomal abnormalities in bone marrow—in particular, the -5, del(5q), -7, del(q) and complex karyotype.
Secondary MDS after treatment with a topoisomerase II inhibitors such as an anthracycline or etoposide occurs 1-3 years after exposure to these agents. The chromosomal abnormalities commonly involve the MLL gene (11q23).
MDS may also develop after exposure to certain chemicals (eg, benzene). Insecticides, weed killers, and fungicides are also possible causes of MDS and secondary leukemia. [4] Viral infections have also been implicated. Less evidence supports genetic predisposition, but familial incidences have been described. Some of the congenital platelet disorders with RUNX1 and GATA2 mutations can predispose to MDS.
Although familial cases of myelodysplastic syndromes are rare, they are immensely valuable for the investigation of the molecular pathogenesis of myelodysplasia in general. [5] The best-characterized familial MDS is familial platelet disorder with propensity to myeloid malignancy, which is caused by heterozygous germline RUNX1 mutations. The incidence of MDS/AML in affected pedigrees is over 40%, with a median age of onset of 33 years. Familial monosomy 7; unusually short telomeres in dyskeratosis congenita; and four pedigrees with inherited MDS caused by heterozygous mutations in GATA2 have been reported. [6] These familial forms may occasionally be found in the course of screening family members of a patient with MDS as bone marrow transplant donors.
A study by Kristinsson et al found that chronic immune stimulation is a trigger for acute leukemia and MDS development. The underlying mechanisms may also be caused by a genetic predisposition or treatment for infections or autoimmune conditions. [7]
Epidemiology
The actual incidence of MDS in the United States is unknown. MDS was first considered a separate disease in 1976, and its occurrence was estimated at 1500 new cases every year. At that time, only patients with less than 5% blasts were considered to have this disorder. MDS was not classified as neoplastic and included in cancer registries until 2001. [8] Current estimates of the incidence of MDS in the United States vary widely, from 10,000 to 30,000-55,000 new cases each year. [8, 9, 10, 11] The higher figures have been questioned as possible overestimates resulting from inclusion of other hematopoietic conditions. [12]
The incidence of MDS has appeared to be increasing. The apparent rise is believed to reflect the increase in the elderly population, but may also reflect improvements in recognition and criteria for the diagnosis. [9]
Although MDS may occur in persons of any age, including children, MDS primarily affects elderly people, with the median onset in the seventh decade of life. Data from 2001 through 2003 of the first National Cancer Institute's Surveillance, Epidemiology & End Reports (SEER) indicate 86% of MDS cases were diagnosed in individuals who were 60 years of age or older (median age: 76y).
Other data from SEER also show that the estimated incidence of MDS increases significantly with age, ranging from 0.7 per 100,000 population during the fourth decade of life to 20.8-36.3/100,000 after age 70 years. There is a fivefold difference in risk between age 60 and ≥80 years.
At all ages, MDS is more common in males than in females. In SEER data from 2001-2003, the incidence rate was significantly higher in men than in women (4.5 vs 2.7 per 100,000 population). [13]
MDS is found worldwide and is similar in characteristics throughout the world. Data based mainly on European numbers from Germany and Sweden were very similar to the US numbers. [11]
A review of United Kingdom population-based data from September 2004 to August 2013 found marked variations in MDS incidence, depending on the standard population used to calculate rates. For example, using the 1996 world standard, the population with the greatest weighting towards younger groups, the incidence rate was 1.67 per 100,000 population; using the 2013 European Standard Population, which has the greatest weighting towards older ages, the rate was 4.4 per 100,000 population. [14]
Prognosis
In some patients, MDS is an indolent disease. Other patients develop significant cytopenias; the resulting complications (eg, bleeding and infections) account for almost all the mortality related to MDS. In the remainder of cases the disease follows an aggressive course and converts into an acute form of leukemia.
Risk classification systems to estimate prognosis in patients with MDS have been developed by the French-American-British (FAB) Cooperative Group, the World Health Organization (WHO), and the MDS Risk Analysis Workshop.
The FAB system classifies MDS into the following five subgroups, differentiating them from acute myeloid leukemia [15] :
-
Refractory anemia (RA)
-
RA with ringed sideroblasts (RARS)
-
RA with excess blasts (RAEB; 6-20% myeloblasts)
-
RAEB in transition to AML (RAEB-T; 21-30% myeloblasts)
-
Chronic myelomonocytic leukemia (CMML)
An underlying trilineage dysplastic change in the bone marrow cells is found in all subtypes.
RA and RARS are characterized by 5% or less myeloblasts in bone marrow. RARS is defined morphologically as having 15% erythroid cells with abnormal ringed sideroblasts (see the image below), reflecting an abnormal accumulation of iron in the mitochondria. Both RA and RARS have a prolonged clinical course and a low prevalence of progression to acute leukemia. In a review of United Kingdom population-based data, with followup of 2 to 11 years, progression to acute leukemia occurred in 5% of RARS cases, compared with 25% of RAEB cases. [14]
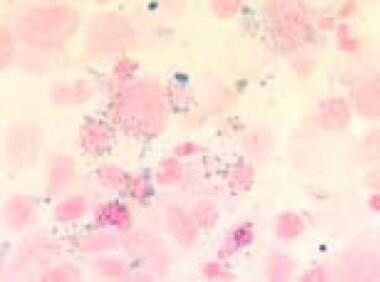 Bone marrow film (1000× magnification) demonstrating ring sideroblasts in Prussian blue staining in a refractory anemia with excess of blasts in transformation. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Bone marrow film (1000× magnification) demonstrating ring sideroblasts in Prussian blue staining in a refractory anemia with excess of blasts in transformation. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
RAEB and RAEB-T (see the image below) are characterized by greater than 5% myeloblasts. The higher the percentage of myeloblasts present, the shorter the clinical course and the closer the disease is to acute myelogenous leukemia.
Blood film (1000× magnification) demonstrating a vacuolated blast in a refractory anemia with excess of blasts in transformation. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Transition from early to more advanced stages may occur, which indicates that these subtypes are merely stages of disease rather than distinct entities. Elderly patients with MDS who progress to acute leukemia are often considered to have a poor prognosis because their disease response to chemotherapy is worse than that of de novo acute myeloid leukemia patients.
The 1999 WHO classification proposed including all cases of RAEB-T in the category of acute leukemia because these patients have similar prognostic outcomes. [16] However, the response to therapy is worse than in patients with de novo or more typical AML or acute nonlymphocytic leukemia.
The fifth type of MDS, CMML, is the most difficult to classify. This subtype can have any percentage of myeloblasts but manifests as a monocytosis of 1000/μL or more, a total white blood cell (WBC) count of less than 13,000/μL, and trilineage dysplasia.
CMML may be associated with splenomegaly. This subtype overlaps with myeloproliferative disease (MPD) and may have an intermediate clinical course. CMML must be differentiated from classic chronic myelocytic leukemia, which is characterized by a negative Ph chromosome.
The current WHO classification lists CMML within a group of myelodysplastic/myeloproliferative neoplasm (MDS/MPN) overlap syndromes, along with four other entities: atypical chronic myeloid leukemia (aCML), BCR-ABL1; juvenile myelomonocytic leukemia (JMML); MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T); and MDS/MPN, unclassifiable. WHO criteria for these forms of CMML include splenomegaly and a WBC count greater than 13,000/μL. [17]
WHO Classification
The WHO classification scheme for MDS was published in 1999. Updates to the scheme were published in 2008 and 2016. The 2016 WHO classification of MDS is as follows [17] :
-
MDS with single-lineage dysplasia (MDS-SLD) – 1 or 2 blood cytopenias; in bone marrow, dysplasia in ≥ 10% of one cell line, < 5% blasts
-
MDS with multilineage dysplasia (MDS-MLD) – 1-3 blood cytopenias, < 1 × 109/L monocytes; in bone marrow, dysplasia in ≥ 10% of cells in ≥ 2 hematopoietic lineages < 15% ring sideroblasts (or < 5% ring sideroblasts if SF3B1 mutation present) < 5% blasts
-
MDS with ring sideroblasts (MDS-RS) – Anemia, no blasts; in bone marrow, ≥ 15% of erythroid precursors with ring sideroblasts or ≥ 5% ring sideroblasts if SF3B1 mutation is present
-
MDS with isolated del(5q) – Anemia, platelets normal or decreased; in bone marrow, unilineage erythroid dysplasia, isolated del(5q), < 5% blasts ± one other abnormality except -7/del(7q)
-
MDS with excess blasts (MDS-EB) – 1-3 blood cytopenias, 0-3 dysplastic bone marrow lineages, and 5-9% blasts in bone marrow or 2-4% blasts in blood (MDS-EB1) or 10-19% blasts in bone marrow or 5-19% blasts in blood (MDS-EB2)
-
Unclassifiable MDS – Cytopenias, ±1% blasts on at least 2 occcasions; in bone marrow, single-lineage dysplasia or no dysplasia but characteristic MDS cytogenetics, < 5% blasts
The WHO classification also includes provisional category of refractory cytopenia of childhood, with cytopenias and < 2% blasts in peripheral blood and, in bone marrow, dysplasia in 1–3 lineages and < 5% blasts.
International Prognostic Scoring System
To improve prognostic classification, the MDS Risk Analysis Workshop developed the Myelodysplastic Syndrome International Prognostic Scoring System (IPSS). The IPSS was published in 1997 and updated in 2012. [18, 19, 20] The revised IPPS (IPSS-R) score is calculated on the basis of five variables:
-
Hemoglobin level
-
Absolute neutrophil count
-
Platelet count
-
Percentage of bone marrow blasts
-
Cytogenetic category
With the IPSS, patients were stratified into four risk groups: low, intermediate 1 and 2, and high. The IPSS-R score is used to stratify patients into five risk groups, as shown in Table 1, below:
Table 1. Revised International Prognostic Scoring System risk groups and prognosis [19] (Open Table in a new window)
Risk Group |
Time to Development of AML (y) |
Median Survival (y) |
Very low |
NR |
8.8 |
Low |
10.8 |
5.3 |
Intermediate |
3.2 |
3.0 |
High |
1.4 |
1.6 |
Very High |
0.7 |
0.8 |
AML – Acute myelogenous leukemia |
||
Mean survival is 18-24 months or longer in patients with the following features:
-
Single or mild cytopenias
-
Normal chromosomes or a single chromosomal abnormality (except those involving chromosome 7)
-
Fewer than 10% myeloblasts in the bone marrow
Mean survival is 6-12 months in patients with the following features:
-
Pancytopenia requiring red blood cell or platelet transfusions
-
Chromosome 7 abnormalities or multiple chromosomal abnormalities
-
Greater than 10% myeloblasts
Personalized Prognostic Model
Using artificial intelligence, Razha and colleagues developed a personalized prognostic model for patients with MDS that is based on clinical and genomic data. [21] Factors identified as having an impact on overall and leukemia-free survivals included the following:
-
Chromosomal karyotype
-
Platelet count
-
Hemoglobin level
-
Bone marrow blast percentage
-
Age
-
Other clinical variables
-
Seven discrete gene mutations (ASXL1, EZH2, KRAS, NRAS, RAD21, SF3B1, and TP53)
-
Number of mutations
Validation of the model confirmed that it outperformed established prognostic models in MDS. The new model was able to predict the probability of survival and of transformation of MDS into leukemia at different time points that are unique for a given patient, permitting the upstaging or downstaging of patients into more appropriate risk categories. [21]
Molecular International Prognostic Scoring System
The International Working Group for the Prognosis of MDS, working under the auspices of the MDS Foundation, has created and validated the Molecular International Prognostic Scoring System for MDS (IPSS-M). This system uses information from 31 gene mutations, along with diagnostic and clinical parameters, to determine a personalized, patient-specific risk score. [22]
The IPSS-M has six risk categories: very low, low, moderate low, moderate high, high, very high). The score can be used for determining clinical trial eligibility criteria, correlative studies, and treatment recommendations. The IPSS-M calculator is available at https://mds-risk-model.com/.
-
Blood film (1000× magnification) demonstrating a vacuolated blast in a refractory anemia with excess of blasts in transformation. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
This bone marrow film (400× magnification) demonstrates an almost complete replacement of normal hematopoiesis by blasts in a refractory anemia with an excess of blasts in transformation. Note the signs of abnormal maturation such as vacuolation, double nucleus, and macrocytosis. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Bone marrow film (1000× magnification) demonstrating ring sideroblasts in Prussian blue staining in a refractory anemia with excess of blasts in transformation. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Bone marrow film (1000× magnification) demonstrating granular and clotlike positive reaction in periodic acid-Schiff staining in a refractory anemia with excess of blasts in transformation. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Tables
- Table 1. Revised International Prognostic Scoring System risk groups and prognosis [19]
- Table 2 Cytogenetic abnormalities assigned an IPSS-R value for scoring
- Table 3.Calculation of IPSS-R score
- Table 4. IPSS-R prognostic risk scores and categories
- Table 5. Clinical outcome by IPSS-R risk category
- Table 6. Categories of FAB classification versus WHO classification for myelodysplastic syndrome (MDS)
- Table. 2008 and 2016 World Health Organization (WHO) classifications of myelodysplastic syndromes
Risk Group |
Time to Development of AML (y) |
Median Survival (y) |
Very low |
NR |
8.8 |
Low |
10.8 |
5.3 |
Intermediate |
3.2 |
3.0 |
High |
1.4 |
1.6 |
Very High |
0.7 |
0.8 |
AML – Acute myelogenous leukemia |
||
Cytogenetic prognostic subgroups |
Cytogenetic abnormalities |
Very good |
-Y, del(11q) |
Good |
Normal, del(5q), del(12p), del(20q), double including del(5q) |
Intermediate |
Del(7q), +8, +19, t(17q), any other single or double independent clones |
Poor |
-7, inv(3)/t(3q)/del(3q), double including -7,/del(7q), complex: 3 abnormalities |
Very poor |
Complex: >3 abnormalities |
Points Assigned |
||||||||
0 |
0.5 |
1 |
1.5 |
2 |
3 |
4 |
||
Variable |
Cytogenetic subgroup |
Very Good |
Good |
Intermediate |
Poor |
Very Poor |
||
Bone marrow blasts (%) |
≤2 |
>2- < 5 |
5-10 |
>10 |
||||
Hemoglobin (g/dL) |
≥10 |
8-9.9 |
< 8 |
|||||
Platelet count (x 109/L) |
≥100 |
50-99.9 |
< 50 |
|||||
Absolute neutrophil count (x 109/L) |
≥0.8 |
< 0.8 |
||||||
Risk Score |
Risk Category |
≤1.5 |
Very Low |
>1.5-3 |
Low |
>3-4.5 |
Intermediate |
>4.5-6 |
High |
>6 |
Very High |
IPSS-R Risk Category |
||||||
Very Low |
Low |
Intermediate |
High |
Very High |
||
Clinical Outcome |
Median survival (years) |
8.8 |
5.3 |
3.0 |
1.6 |
0.8 |
Median time to 25% acute myelogenous leukemia evolution (years) |
NR |
10.8 |
3.2 |
1.4 |
0.7 |
|
FAB Classification |
WHO–2004 Classification |
WHO–2008 Classification |
RA |
RA RCMD 5q- |
RCUD RCMD 5q- |
RARS |
RARS RCMD-RS |
RARS RCMD-RS RARS-T |
RAEB |
RAEB-1 RAEB-2 |
RAEB-1 RAEB-2 |
CMML |
CMML-1 CMML-2 |
CMML-1 CMML-2 |
RAEB-T |
AML |
AML |
| 2008 WHO Classification [17] | 2016 WHO Classification [52] |
| Refractory cytopenia with unilineage dysplasia (RCUD) encompassing refractory anemia (RA), refractory neutropenia (RN), and refractory thrombocytopenia (RT) | MDS with single-lineage dysplasia (MDS-SLD) |
| Refractory cytopenia with multilineage dysplasia (RCMD) | MDS with multilineage dysplasia (MDS-MLD) |
| Refractory anemia with ringed sideroblasts (RARS) | MDS with ring sideroblasts (MDS-RS) MDS-RS with single lineage dysplasia (MDS-RS-SLD) MDS-RS with multilineage dysplasia (MDS-RS-MLD) |
| Myelodysplastic syndrome associated with isolated del(5q) | MDS with isolated del(5q) |
| MDS with excess blasts (MDS-EB) | |
| Refractory anemia with excess blasts–1 (RAEB-1) | MDS-EB-1 |
| Refractory anemia with excess blasts–2 (RAEB-2) | MDS-EB-2 |
| Myelodysplastic syndrome, unclassified (MDS-U) | MDS, unclassifiable (MDS-U) with 1% blood blasts with single lineage dysplasia and pancytopenia based on defining cytogenetic abnormality |
| Refractory cytopenia of childhood | Refractory cytopenia of childhood |
