Practice Essentials
Pearson marrow-pancreas syndrome, an often fatal disorder, was first described in 1979, by pediatric hematologist/oncologist Howard Pearson. Affected infants manifest a refractory, transfusion-dependent sideroblastic anemia; vacuolization of hematopoietic precursors; and exocrine pancreatic insufficiency. [1] The last, although frequently encountered, may be absent in some cases of Pearson syndrome. The condition is now known to be a rare, multisystemic, mitochondrial cytopathy with anemia, neutropenia, and thrombocytopenia, as well as variable hepatic, renal, and endocrine failure. Death usually occurs early in life (before age 4 years). The most common causes of death are lactic acidemia (which may be triggered by infection) and liver or renal failure. [2] Survivors after early childhood develop features of Kearns-Sayre syndrome (KSS), a mitochondriopathy characterized by progressive external ophthalmoplegia, weakness of skeletal muscle, atypical retinal pigmentation, and cardiac conduction defects. Patients may recover from the refractory anemia.
In adulthood a phenotypically milder disorder, chronic progressive external ophthalmoplegia (CPEO), is seen; it is marked by variable ptosis, ophthalmoplegia, and proximal limb weakness.
Pearson syndrome is due to mitochondrial DNA (mtDNA) deletions of variable size and location; mtDNA encodes for 13 of the respiratory chain enzymes, along with 24 RNA molecules used in intramitrochondrial protein synthesis. As a result, defective oxidative phosphorylation, as well as other defects (occurring in enzymes and RNA molecules), is involved in the syndrome's etiology.
Signs and symptoms of Pearson syndrome
Birth weight may have been low, and the infant may not have gained weight well. This may be confirmed with a careful growth chart.
Chronic diarrhea and fatty stools may be noted and suggest pancreatic exocrine deficiency as a cause for failure to thrive.
No pathognomonic physical characteristics are observed. Anemia causes pallor, and the patient's weight may be low for the person’s age.
Workup in Pearson syndrome
Laboratory studies include the following:
-
Complete blood count (CBC) with differential and reticulocyte count - Patients with Pearson syndrome have macrocytic anemia; the reticulocyte count is inappropriately low for the degree of anemia; some patients also have leukopenia, neutropenia, and/or thrombocytopenia
-
Pancreatic exocrine function testing
-
Serum lactic acid measurement - Patients may have lactic acidemia, most commonly seen during intercurrent illnesses
-
Urinalysis - Complex organic aciduria, including 3-methylglutaconic aciduria, has been reported [3]
-
Hepatic study - Hepatic transaminase values may be increased in patients with hepatic involvement; bilirubin levels may be increased, and albumin concentrations and coagulation values (eg, prothrombin time) may reflect a defect in synthetic function
-
Endocrinologic study - Some patients have evidence of thyroid, parathyroid, or growth hormone deficiency
-
Analysis of mitochondrial DNA
Bone marrow aspiration and biopsy are necessary to obtain bone marrow for histologic analysis. The number of erythroid precursors in the bone marrow is normal or increased, and there is a characteristic vacuolization of hematopoietic precursors.
Management of Pearson syndrome
No specific therapy is available for individuals with Pearson syndrome or other mitochondrial cytopathies. Red blood cell transfusions are often needed to manage the macrocytic anemia, and patients may be dependent on transfusions. Pancreatic enzyme replacement is needed for patients with malabsorption due to exocrine pancreatic insufficiency. Supplementation with fat-soluble vitamins may also be needed.
Parenteral antibiotics should be administered after blood for culture is obtained, for any fever over 101.5ºF (38.6ºC).
Intermittent metabolic crises are managed with supportive care, including hydration, correction of electrolyte abnormalities or acidosis, and treatment of underlying causes (eg, infection). Evidence of concomitant hepatic failure should be sought.
Patients may have hypothyroidism, hypoparathyroidism, diabetes mellitus, or growth hormone deficiency. These conditions should be screened for and treated, if present.
Pathophysiology
Mitochondriopathies
The mitochondriopathies comprise several diverse, overlapping syndromes caused by mutations of mitochondrial DNA. [4] Pearson syndrome is a specific clinical subset of these syndromes in which involvement of the bone marrow and exocrine pancreas is prominent. The pathogenesis of Pearson syndrome is complex and not well understood. Deletions of certain components of the electron transport chain, encoded by mitochondrial DNA, cause a defect in cellular oxidative metabolism. Certain transfer RNAs (tRNAs) may also be deleted, and their deletion impairs the translation of messenger RNAs (mRNAs) to proteins.
The specific mtDNA deletion includes deletion of the complete genes for ATPases 6 and 8, cytochrome c oxydase III, and NADH dehydrogenase 3, 4, 4L, and 5. [5, 6, 7, 8]
These defects cause cellular injury in target tissues.
Other mitochondriopathies, such as Kearns-Sayre syndrome and the mitochondrial myopathies, have deletions of mitochondrial DNA that may be similar or identical to those detected in Pearson syndrome. How similar abnormalities of mitochondrial DNA cause such diverse disorders is not well understood. The distinct phenotypes may be the result of differences in the amount and in the tissue-specific distribution of abnormal mitochondrial DNA, the evolution of this distribution over time, and the effects of tissue-specific nuclear modifier genes. [9, 10]
Whether mitochondrial DNA deletion size and location are associated with the severity of Pearson syndrome remains unclear. In four patients with the disease studied by Son et al, deletion size did not strongly correlate with phenotype. However, earlier symptom onset occurred in a patient with longer deletions. [11]
Defining features of Pearson syndrome
The first defining feature of Pearson syndrome is marrow failure. Sideroblastic anemia, usually macrocytic and frequently transfusion dependent, is observed in isolation or associated with neutropenia and thrombocytopenia. Bone marrow examination is hypoplastic with characteristic vacuolation of hematopoietic precursors and ringed sideroblasts (see the images below).
In the study by Son et al of four patients with Pearson syndrome, the investigators found that the severity of bone marrow dysfunction varied from pancytopenia to transient anemia. [11]
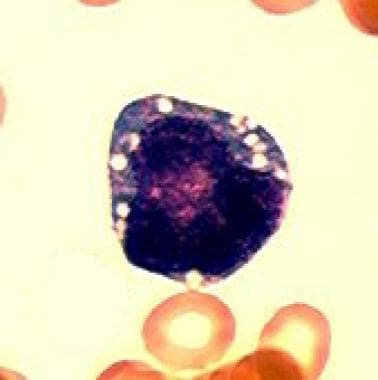 Characteristic vacuolization of a hematopoietic precursor in the bone marrow. (Light microscopy; 100x; Wright-Giemsa stain)
Characteristic vacuolization of a hematopoietic precursor in the bone marrow. (Light microscopy; 100x; Wright-Giemsa stain)
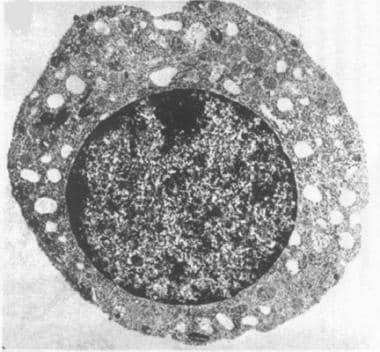 Electron photomicrograph of a hematopoietic precursor (normoblast) with vacuolization. (Transmission electron microscopy; original 10,000x)
Electron photomicrograph of a hematopoietic precursor (normoblast) with vacuolization. (Transmission electron microscopy; original 10,000x)
The second defining feature of Pearson syndrome is dysfunction of the exocrine pancreas due to fibrosis and acinar atrophy. The result is malabsorption, chronic diarrhea, and poor growth or failure to thrive. Studies have reported that 23-63% of patients with Pearson syndrome do not suffer from pancreatic insufficiency. In a retrospective cohort study by the Marrow Failure Study Group of the Associazione Italiana Emato-Oncologia Pediatrica (AIEOP), the investigators found no such insufficiency in 73% of patients. [12]
Another cardinal feature of Pearson syndrome is persistent or intermittent lactic acidemia, which is caused by defects in oxidative phosphorylation. Increased lactate/pyruvate ratio is observed along with increased urinary excretion of lactate and related organic acids.
Other organ systems are affected in various ways. Hepatic involvement may cause increases in transaminase, bilirubin, and lipid levels, as well as in steatosis. Some patients develop hepatic failure. Renal involvement is common and manifests as a tubulopathy, such as Fanconi syndrome. Endocrinologic disturbances, such as growth hormone deficiency, hypothyroidism, and hypoparathyroidism, may develop but are not usually part of the initial presentation. [13] The endocrine pancreas usually remains functional; however, historically, a few patients develop diabetes mellitus and adrenal insufficiency. Splenic atrophy and impaired cardiac function have also been reported. [14, 15, 16, 17, 18]
Siri et al reported a systematic, single-center evaluation of 18 persons with single, large-scale mitochondrial DNA deletions (6 with Pearson syndrome and 12 with KSS) in which nearly half developed evidence of adrenocortical dysfunction. With provocative testing, 75% of persons with Pearson syndrome had adrenal insufficiency by age 3 years, while KSS patients developed subclinical adrenocortical dysfunction at a median of 6.6 years. The investigators suggested the need for ongoing systematic testing for endocrine conditions in children with these disorders. [19]
Failure to thrive is common. Several factors are likely contributory. Such factors include a defect in cellular metabolic energy, malabsorption due to exocrine pancreatic failure, hepatic and renal insufficiency, abnormal myelinization, and possibly endocrinologic abnormalities. [20, 21]
Epidemiology
Frequency
United States
Pearson syndrome is rare. Fewer than 1000 cases have been reported worldwide.
International
See United States.
Mortality/Morbidity
Pearson syndrome is often fatal in infancy or early childhood. The usual causes of death are bacterial sepsis due to neutropenia, metabolic crisis, and hepatic failure.
A retrospective study by Anteneová et al of patients with single, large-scale mitochondrial DNA deletions found that of the five with Pearson syndrome, 5-year survival was 60%, compared with 100% for patients with other mitochondrial cytopathies such as progressive external ophthalmoplegia and Kearns-Sayre syndrome spectrum. [22]
Race
All races can be affected.
Sex
Pearson syndrome has no sex predilection.
Age
Pearson syndrome is a progressive disease, and its features change with age. Neonates may be well at birth, but some 40% of patients present in the first year with persistent hypoplastic macrocytic anemia, other cytopenias, low birth weight/poor weight gain, microcephaly, and multiple organ system involvement (GI, neuromuscular, and metabolic). [20, 23, 24] Hydrops fetalis has also been reported. Anemic newborns may need transfusion.
In the aforementioned study by Anteneová et al, of patients with single, large-scale mitochondrial DNA deletions, it was found that all of the patients with Pearson syndrome manifested the condition before age 1 year, with 80% of them presenting with transfusion-dependent sideroblastic anemia. [22]
During infancy and early childhood, failure to thrive, chronic diarrhea, and progressive hepatomegaly often occur in individuals with Pearson syndrome. These conditions are punctuated by episodic metabolic crises characterized by somnolence, vomiting, electrolytic abnormalities, lactic acidosis (elevated lactate:pyruvate ratio), and hepatic insufficiency. The metabolic crises are often precipitated by infection. The lactic acidosis may become persistent with time. Typical causes of death in infants and young children with Pearson syndrome are metabolic crisis, hepatic failure, and overwhelming sepsis related to neutropenia.
Some patients survive infancy and early childhood and spontaneously recover from the hematologic dysfunction. Case reports document a shift in the phenotype of these individuals to a predominantly myopathic or encephalopathic condition. For example, some patients who survive early childhood may develop Kearns-Sayre syndrome or Leigh syndrome, whereas others may be neurologically healthy. In the aforementioned AIEOP study, the investigators reported that, while all of the 11 patients in the study tested neurologically normal at birth, seven of them (64%) subsequently suffered from retardation of speech development, hypotonia, and muscle hypotrophy, with three patients eventually approaching a complete Kearns-Sayre syndrome phenotype. [12]
-
Characteristic vacuolization of a hematopoietic precursor in the bone marrow. (Light microscopy; 100x; Wright-Giemsa stain)
-
Electron photomicrograph of a hematopoietic precursor (normoblast) with vacuolization. (Transmission electron microscopy; original 10,000x)
-
Ringed sideroblast in the bone marrow (iron stain). The dark structures that form a ring around the nucleus are hemosiderin-laden mitochondria. (Light microscopy; 100x; iron stain)
Tables

What would you like to print?
- Exocrine Pancreatic Insufficiency
- Fast Five Quiz: Exocrine Pancreatic Insufficiency
- Fast Five Quiz: Exocrine Pancreatic Insufficiency Presentation and Diagnosis
- Fast Five Quiz: Exocrine Pancreatic Insufficiency Management
- Fast Five Quiz: Exocrine Pancreatic Insufficiency Signs and Symptoms
- A Coffee Drinker in Severe Pain
- Diagnosis and Management of Exocrine Pancreatic Insufficiency
- Exocrine Pancreatic Insufficiency (EPI): 5 Things to Know
- Exocrine Pancreatic Insufficiency: Optimal PERT Dose Varies by Primary Pancreatic Disease
-
 Cause of Common Gastrointestinal Symptoms in Diabetes?
Cause of Common Gastrointestinal Symptoms in Diabetes?
-
Busting Some Popular Myths About Exocrine Pancreatic Insufficiency
-
High Cost of Pancreatic Enzymes a Barrier for Patients With Cancer




