Practice Essentials
Phenylketonuria (PKU), less commonly known as phenylalanine hydroxylase (PAH) deficiency, is the most common inborn error of amino acid metabolism, arising from bi-allelic pathogenic variants in the gene that encodes PAH. [1, 2] For the sake of familiarity, the terms PKU and phenylketonuria will be used interchangably in this article. A deficiency of the enzyme phenylalanine hydroxylase (PAH) impairs the body’s ability to metabolize the essential amino acid phenylalanine into tyrosine in the proximal step of the metabolic pathway. This leads to accumulation of phenylalanine in body fluids, and decreased tyrosine. In plasma, normal phenylalanine level is less than 120 umol/L (2 mg/dL), and in untreated patients with PKU, levels can be significantly above this threshold, often above 1000 umol/L. [3]
Persistently elevated phenylalanine levels lead to profound and irreversible intellectual disability; other disease manifestations across the lifespan in an untreated individual may include epilepsy, eczema, musty body odor, behavioral issues, decreased skin and hair pigmentation, and Parkinson-like features. It is also worth noting that elevated Phe levels are teratogenic. With appropriate treatment and careful protein restriction from early infancy, individuals with PKU develop typical intellectual functioning. [1]
Signs and symptoms
Skin findings in PKU are as follows:
-
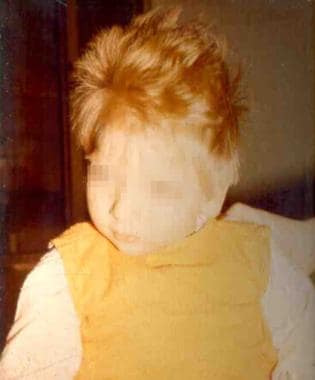
Hypopigmented skin and hair [1, 3] : The inability to metabolize phenylalanine leads to low tyrosine levels and inhbition of the tyrosinase enzyme. Tyrosine is a precursor of melanin. Untreated PKU therefore impacts melanin synthesis and leads to hypopigmentation, the most characteristic cutaneous manifestation of PKU. It can be striking in black and Japanese patients, although not all untreated patients are fair; treated patients often have typical pigmentation
-
Eczema (including atopic dermatitis)
-
Light sensitivity
-
Increased incidence of pyogenic infections
-
Increased incidence of keratosis pilaris
-
Decreased number of pigmented nevi
-
Sclerodermalike plaques
-
Hair loss [4]
Other manifestations of untreated PKU are as follows [1, 3] :
-
Intellectual disability (the most common finding overall)
-
Musty or mousy odor
-
Epilepsy
-
Extrapyramidal manifestations (eg, parkinsonism)
-
Progressive white matter disease on brain MRI
-
Mood disorder
Diagnosis
Screening for PKU involves the following [1, 3, 5] :
-
Determination of phenylalanine levels: The standard amino acid analysis done by means of ion exchange chromatography or tandem mass spectrometry. In the United States, this is part of routine newborn screening.
-
The Guthrie test as a bacterial inhibition assay: Formerly used, but now being replaced by tandem mass spectrometry
-
Molecular testing is generally unnecessary for a diagnosis of PKU; given that it is autosomal recessive in nature, bi-allelic pathogenic variants are requried to establish the diagnosis. To date, more than 900 pathogenic variants have been described in PAH (see www.biopku.org)
Imaging studies
Cranial magnetic resonance imaging (MRI) studies may be indicated in older individuals who have abandoned the diet used to manage PKU and are experiencing deficits in motor or cognitive function, or in cases in which behavioral, cognitive, or psychiatric concerns exist. Prolonged exposure to elevated phenylalanine levels has been found to be detrimental to white matter integrity. [6] In terms of volume loss, the most severely affected brain structures are the cerebrum, the corpus callosum, the hippocampus, and the pons. [7]
Management
Dietary management and/or pharmacologic treatment are essential for patients with PKU. [1, 3]
Dietary treatment
The mainstay of dietary management for patients with PKU consists of phenylalanine restriction which is accomplished by natural protein restriction, as well as the use of medical foods (commonly known as 'metabolic formula') to supplement the patient’s intake of other essential amino acids and of vitamins and minerals. [1, 8] Energy and variety are provided by low-protein foods, including fruits, nonstarchy vegetables, and specially ordered low-protein items. Agents to avoid in the diet, other than excess natural protein which may contain high Phenylalanine levels, include aspartate, an artificial sweetner that contains phenylalanine.
Pharmacologic management
Sapropterin (Kuvan) is a synthetic form of BH4, the cofactor for the enzyme PAH. It may reduce phenylalanine levels or improve natural protein tolerance in patients who are deemed sapropterin-responsive, but it does not substitute the need for a metabolic diet. [2, 9]
Pegvaliase (Palynziq) is a PEGylated phenylalanine ammonia lyase (PAL), which was FDA approved in May 2018. It is administred via subcutaneous injection, and exerts effect by converting phenylalanine to trans-cinnamic acid. In patients in whom it is effective, it can eliminate the need for protein restriction. [10]
Patients who have suboptimal dietary treatment may benefit to some degree from consuming large neutral amino acids, which may block phenylalanine entry into the brain and may also result in a modest lowering of plasma phenylalanine levels.
Background
Phenylalanine hydroxylase (PAH) deficiency, better known as PKU, is the most common inborn error of amino acid metabolism. It results from an impaired ability to metabolize the essential amino acid phenylalanine, leading to accumulation in blood and brain. [1, 3]
Several different classifications have been used in the past to describe PKU severity. Commonly, classic PKU is considered to be present when untreated plasma phenylalanine levels exceed 20 mg/dL (1200 µmol/L) without treatment. Milder degrees of plasma phenylalanine elevation are often referred to as hyperphenylalaninemia. The American College of Medical Genetics and Genomics has released a practice guideline acknowledging that PAH deficiency represents an unbroken spectrum of disease. [2]
Persistently elevated phenylalanine levels negatively impact cognitive function, and untreated individuals with classic PKU may develop profound and irreversible intellectual disability. While treatment is of particular importance in the first few years of life, as childhood brain and pysychomotor development are in a period of initial growth, adolescent or adult patients who go off treatment may also begin to manifest executive functioning difficulties, mood disorder, or white matter disease. [3]
In the United States and many other countries, PKU is detected by newborn screening, and individuals who are appropriately treated (eg, with a diet low in phenylalanine and/or tetrahydrobiopterin) can have typical intelligence and lead a largely typical life.
Pathophysiology
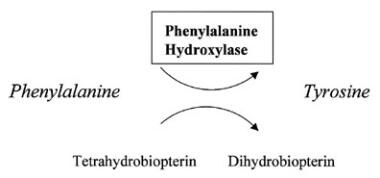
In most patients, the classic type of PKU involves a deficiency of PAH that leads to increased levels of phenylalanine in the plasma (>1200 µmol/L; reference range, 35-120 µmol/L), Phenylalanine hydroxylase catalyzes the conversion of L-phenylalanine to L-tyrosine, the rate-limiting step in the oxidative degradation of phenylalanine. [3] This leads to accumulation of phenylalanine, which is transaminated to phenylpyruvate and other phenylketones. Tyrosine synthesis is therefore impaired, and so tyrosine, a precursor of melanin and catecholamines, is often low, without appropriate dietary supplementation and medical management.
The enzyme PAH crystallizes as a tetramer, with each monomer consisting of a catalytic domain and a tetramerization domain. Examination of the mutations causing PKU reveals that some of the most frequent mutations are located at the interface of the catalytic and tetramerization domains.
The mechanism by which elevated phenylalanine levels cause intellectual disability is not known, although restriction of dietary phenylalanine ameliorates this effect if initiated within a few weeks of birth. A strong relation between control of blood phenylalanine levels in childhood and intelligence quotient (IQ) is recognized.
Subtle neuropsychological deficits in children with treated PKU are under investigation. Some investigators have attributed these deficits to small residual neurotransmitter abnormalities (eg, reduced production of neurotransmitters as a result of deficient tyrosine transport across the neuronal cell membranes).
Phenylalanine hydroxylase requires a nonprotein cofactor termed tetrahydrobiopterin (BH4). A small percentage of children with elevated phenylalanine levels exhibit normal PAH levels/function but have a deficiency in synthesis or recycling of BH4 known as tetrahydrobiopterin deficiency. This condition is in the differential diagnosis of elevated Phe levels in a new patient, and can result from biallelic mutations in the GCH1, PCB1, PTS, or QDPR genes. The BH4 cofactor is also required for hydroxylation of tyrosine (a precursor of dopamine) and tryptophan (a precursor of serotonin). Thus, individuals with BH4 cofactor deficiency can have additional neurologic problems that are not fully corrected by dietary phenylalanine reduction alone, but often require additional treatments that may not be fully effective. In the modern day era, genetic work up following an elevated Phe level that is most often detected by newborn screening includes screening for biopterin defects.
Etiology
Phenylketonuria (PKU) is an autosomal recessive disorder caused by pathogenic variants in the PAH gene, which expresses PAH. [3] The previous term, 'mutation,' has largely been replaced with the term 'pathogenic variant.' The PAH gene is located on 12q23.2, spans about 171 kb, and contains 13 exons. More than 900 different mutations in the PAH gene have been identified to date, and www.biopku.org is a growing resource for genotype-phenotype correlations or descriptions.
The PAH gene shows great allelic variation, and pathogenic mutations have been described in all 13 exons of the PAH gene and its flanking region. The mutations can be of various types, including missense mutations (62% of PAH alleles), small or large deletions (13%), splicing defects (11%), silent polymorphisms (6%), nonsense mutations (5%), and insertions (2%). [11]
The 3 genes related to biopterin synthesis defects are located at 11q22.3-23.3, 10q22, and 2p13, and the gene for biopterin recycling defects is located at 4p15.1-16.1.
Phenylketonuria displays a marked genotypic heterogeneity, both within populations and between different populations. There is some broad genotype-phenotype correlation (alleles that tend to be severe and alleles that tend to be mild), but as is often the case with inborn errors of metabolism, even siblings with the same pathogenic variants and similar broad genetic background may have varying degrees of disease manifestation and severity.
Epidemiology
The frequency of PKU as reported by newborn screening programs varies between one in 13,500 to 19,000 newborns in the United States. While PKU can impact individuals from diverse ethnic backgrounds, it is less common in Americans of African descent and Ashkenazi Jews.
United States statistics
The frequency of PKU as reported by newborn screening programs varies between one in 13,500 to 19,000 newborns in the United States. While PKU can impact individuals from diverse ethnic backgrounds, it is less common in Americans of African descent and Ashkenazi Jews. [12]
International statistics
Phenylketonuria (PKU) frequency varies across populations, [1] with a range of occurrences reported worldwide. In Turkey and Ireland, the frequency is more than 1 in 5000, while individuals of northern European and East Asian origin have a frequency of approximately 1 in 10,000. Specific high incidences have been documented in various regions, including Turkey (approximately 1 case in 2600 births), Yemenite Jewish population (1 in 5300), Scotland (1 in 5300), Estonia (1 in 8090), [13] Hungary (1 in 11,000), and several other countries. Conversely, Finland reports a low incidence of less than 1 in 100,000, [14] along with Japan at 1 in 125,000. [11]
Age-, sex-, and race-related demographics
Phenylketonuria most commonly is diagnosed in neonates because of universal newborn screening programs. However, for individuals born before the advent of universal newborn screening, or born in countries where newborn screening for PKU is not established, PKU can present in an individual of any age and background.
PKU can occur in either sex with equal frequency.
Elevated phenylalanine levels are highly teratogenic. People with PKU who are pregnant must be monitored closely during pregnancy to maintain Phe levels within the recommended therapeutic range of 120-360 umol/L. Persistnetly elevated Phe levels during pregnancy has been associated with microcephaly, brain malformations, limb malformations, congenital heart defect, or other congenital anomalies.
Prognosis
Early initiation of treatment for phenylketonuria (PKU) in the first few days of life is critical in preventing the severe manifestations of the disease. [2] While a strict low-phenylalanine diet can help manage symptoms, mild cognitive deficits and mental health issues may still arise even with good dietary control. [15, 16]
If treatment is not started until after 2 to 3 years of age, it may only effectively control extreme hyperactivity and seizures, with limited impact on cognitive outcomes. Children born to mothers with poorly controlled PKU during pregnancy are at high risk of microcephaly and developmental deficits in utero.
The prognosis for normal intelligence is excellent when patients are promptly placed on a low-phenylalanine diet within the first month of life and receive careful and frequent monitoring. [17] Full management by a medical biochemical geneticist and an experienced metabolic dietitian is essential for successful outcomes. [1]
However, some children may experience mild impairments in school functioning, particularly when dietary control is not optimal. Regular follow-up visits and adherence to dietary guidelines are necessary to optimize long-term health and cognitive outcomes for individuals with PKU. [1]
Patient Education
Teach parents how to administer the diet at home, and count grams of natural protein in foods, and involve all caregivers as well. Children should begin involvement in their dietary planning as soon as they are developmentally ready. Poor dietary control is often associated with increasing noncompliance by older children, but it could also be due to a more relaxed dietary approach by parents and increasing dietary errors. [18]
Women with PKU should be educated about the risks of untreated pregnancy and the benefits of dietary and, in some cases, pharmacologic, treatment. [2] Patients with PKU should avoid aspartame (an artificial sweetener). Aspartame is widely used in medicines, vitamins, beverages, and other substances.
The phenylalanine-restricted diet with semisynthetic supplementation is not without risk. Patients with PKU under dietary treatment can have low concentrations of trace elements and cholesterol and can have some disturbance to folate metabolism and distortion of their fatty acid profile. [11] Patients may also have decreased intake of calcium, vitamin D, and vitamin B12. However, much of this is mitigated when under the guidance of an experienced metabolic dietitian.
The organization National PKU News is a nonprofit entity dedicated to providing accurate news and information to families and professionals dealing with PKU. This site contains excellent articles and links to other information sources. Information on how to subscribe to a PKU newsletter and on how to contact support groups is available. Numerous other PKU Web sites are available to assist families in search of additional information.
-
Phenylalanine hydroxylase converts phenylalanine to tyrosine.
-
Fair skin and hair resulting from impairment of melanin synthesis.
