Practice Essentials
Childhood polyarteritis nodosa (CPAN) is a rare and often fatal disease that affects small and medium arteries. [1] (See the image below.) Previously, it had been referred to as infantile polyarteritis nodosa (IPAN). The use of the term infantile is too restrictive, as infancy connotes age 1 year or younger. Polyarteritis has been described worldwide, although vasculitic diseases tend to be more common in individuals of Asian descent.
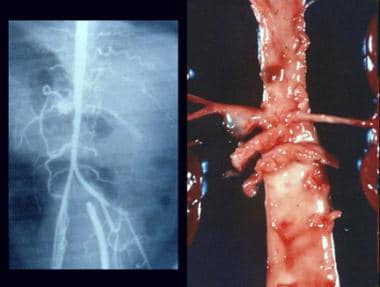 Childhood polyarteritis nodosa. Multiple aneurysms on arteriogram and autopsy specimen.
Childhood polyarteritis nodosa. Multiple aneurysms on arteriogram and autopsy specimen.
Clinically, CPAN often is part of the spectrum of Kawasaki disease (KD). However, it was described nearly 130 years ago. CPAN with aneurysmal involvement of major coronary arteries and KD are clinically and pathologically indistinguishable. Indeed, the major distinction between KD and CPAN is that the diagnosis of KD is based entirely on clinical criteria, whereas the diagnosis of CPAN is based on histologic findings. This article explores the similarities and differences between these entities, with the focus on the current understanding of CPAN.
Signs and symptoms
Presenting symptoms of CPAN are nonspecific and include fever, malaise, anorexia, weight loss, and abdominal pain. In decreasing order of frequency, the organs most often affected are the kidney, heart, and liver.
See Presentation for more detail.
Diagnosis
Laboratory studies
As with other systemic rheumatic diseases, the basic laboratory tests in childhood polyarteritis nodosa (CPAN) should include the following:
-
Complete blood cell (CBC) count
-
Urinalysis
-
Serum chemistry panel
-
Erythrocyte sedimentation rate
-
Antinuclear antibody determination
-
Rheumatoid factor test and tests for hepatitis B
-
Quantitative immunoglobulins
-
Tests for circulating immune complexes (eg, Raji cell radioimmune assay, C1q binding test)
Other tests are selected on the basis of the clinical examination.
Imaging studies
After a basic posteroanterior (PA) and lateral chest radiograph, imaging studies are directed at involved organ systems
See Workup for more detail.
Management
Medical care of the patient with CPAN is individualized. Often, corticosteroids are required to reduce inflammation.
See Treatment and Medication for more detail.
Patient education
Patient and parent education must be individualized and coordinated by a multidisciplinary team, including a nurse, social worker, occupational therapist, physical therapist, and rheumatologist.
Background
The Viennese pathologist Karl von Rokitansky is credited with the first description of polyarteritis nodosa (PAN) in 1852. The drawing that accompanied his original description of PAN clearly showed multiple aneurysms of varying size in the mesenteric artery of the index case. In 1866, Kussmaul and Maier reported the case of a 27-year-old man who, over a period of approximately 2 months, developed a multisystem disease characterized by fever, myalgias, abdominal pain, mononeuritis multiplex, and proteinuria. A few days before his death he developed palpable subcutaneous nodules. At autopsy, nodules involving the coronary, gastric, renal, splenic, mesenteric, hepatic, bronchial, and phrenic arteries were obvious.
Microscopic studies demonstrated that the intima of the affected arteries was completely intact and that the media and adventitia were severely inflamed and disrupted. For these reasons, Kussmaul and Maier termed this condition periarteritis nodosa. Their paper included a drawing of their patient's heart showing numerous coronary artery aneurysms. The first case of CPAN reported in the English-language literature may be that of a 7-year-old boy who, in 1870, died of "scarlet fever" at St. Bartholomew Hospital in London. Samuel Gee noted that 3 coronary artery aneurysms were present at autopsy and were filled with fresh thrombi.
More recently, Sarah Long suggested that perhaps one of the first documented cases of KD in the United States was that of a 9-month-old infant reported as 1 in a series of 5 cases of Stevens-Johnson syndrome in the Journal of Pediatrics in 1949. The infant had fever, irritability, cervical adenopathy, a polymorphous rash, and conjunctival suffusion. Cardiac arrest supervened, and, at autopsy, hemopericardium secondary to a ruptured coronary artery aneurysm was discovered. This author submits that this infant had CPAN. Other similar case reports appeared in Europe and the United States in the late 19th and early 20th centuries.
Much has been written in the past 2 decades with regard to the classification of the systemic vasculitides. In an attempt to put PAN into proper perspective, the classification promulgated by an international consensus conference held in Chapel Hill, NC, and published in 1994 [2] is included here. Large-vessel vasculitis includes giant cell (temporal) arteritis and Takayasu arteritis. Medium-vessel vasculitis includes PAN and KD. Small-vessel vasculitis includes Wegener granulomatosis, Churg-Strauss syndrome, microscopic polyangiitis (microscopic polyarteritis), Henoch-Schönlein purpura, essential cryoglobulinemic purpura, and cutaneous leukocytoclastic angiitis.
Etiology
An infectious etiology for polyarteritis nodosa (PAN) has been considered for years. Early observers considered streptococci or Staphylococcus aureus to be likely candidates. In 1970, Gocke et al [3] demonstrated Australia antigen (hepatitis B surface antigen) and immunoglobulin M (IgM) antibody in an arterial lesion of PAN in a woman who had been transfused with contaminated blood several weeks previously. These remarkable and serendipitous observations by Gocke et al clearly linked hepatitis B surface antigen to the etiology of PAN. Hepatitis B surface antigenemia is associated with approximately 20% of patients with PAN. [4]
Other investigators have suggested various bacterial etiologies, but none has been confirmed independently. Viruses other than hepatitis B, such as hepatitis A and C, human immunodeficiency virus (HIV), cytomegalovirus (CMV), human T-cell lymphotropic virus-1 (HTLV-1), and parvovirus, have been reported to have etiologic associations with PAN, but none has been repeatedly isolated from patients with PAN.
Considerable evidence supports the notion that PAN is an immune complex disease. However, the elusive antigen or antigens involved remain unknown, with the exception of hepatitis B.
The cause of childhood polyarteritis nodosa (CPAN) is not known. Almost since Kawasaki first described acute febrile mucocutaneous lymph node syndrome (MCLNS) of childhood, investigators have attempted to link it with infectious agents or antigens. Rickettsia species, Propionibacterium acnes, a feline virus, retroviruses, Epstein-Barr virus (EBV), a dust mite antigen, streptococci, and a "super antigen" were proposed as etiologic; however, no explanation has stood the test of time. Clinically, MCLNS has features of an infectious disease, perhaps viral. Similarly, it has many features of a rheumatic disease.
Taken together, the evidence suggests that MCLNS/Kawasaki disease (KD) is the final common clinical pathway resulting from any of a number of infecting or inciting agents or antigens. The author has treated 2 children with CPAN and hepatitis B surface antigenemia.
Epidemiology
United States statistics
The incidence and prevalence of childhood polyarteritis nodosa (CPAN) is not known, perhaps because of problems with the nosology of vasculitis syndromes, the rarity of the condition, and the lack of reported cases. The incidence of classic Kawasaki disease (KD) seems to be decreasing in the United States, as well as worldwide, with atypical or forme fruste cases becoming more common.
International statistics
The incidence, prevalence, and distribution of childhood polyarteritis nodosa (CPAN) are not known.
A population-based study from southern Sweden reported that the annual incidence for polyarteritis nodosa was 0.7 per million children. [5]
Race-, sex-, and age-related demographics
Race
Sufficient worldwide data are lacking, but individuals of Asian descent appear to have a disproportionately high incidence of vasculitis.
Sex
Males are affected more commonly than females, with a male-to-female ratio approaching 2:1.
Age
Polyarteritis nodosa (PAN) most often affects persons aged 40-60 years, although all ages are represented. By definition, childhood polyarteritis nodosa (CPAN) refers to cases occurring in childhood.
Prognosis
Overall prognosis is guarded. The 10-year mortality rate, even in aggressively treated patients, exceeds 20%. However, some cases remit without treatment. These statistics relate primarily to adult polyarteritis nodosa (PAN) and not childhood polyarteritis nodosa (CPAN).
Morbidity/mortality
Adult polyarteritis nodosa (PAN) is an often fatal disease. Before the modern treatment era, the 5-year mortality was approximately 90%. The disease course is highly variable in individual patients. Some may have rapidly progressive disease, leading to death in days or weeks, whereas others may have more subacute disease. Other patients experience waxing and waning of symptoms, leading to chronic disability. In still others, the disease apparently remits with little or no treatment.
Childhood polyarteritis nodosa (CPAN) is perhaps more variable; however, rapidly progressive cases involving the coronary arteries may be highly lethal. With the widespread recognition of Kawasaki disease (KD) and its effective treatment, CPAN has almost disappeared. Most cases of isolated coronary arteritis observed today are considered incorrectly (in the author's opinion) to be atypical KD.
Complications
Complications are related to the extent of disease and include digital necrosis and amputation, bowel infarction, myocardial infarction, stroke, renal failure, hepatic failure, and death. (See the image below.) These complications relate primarily to adult polyarteritis nodosa (PAN).
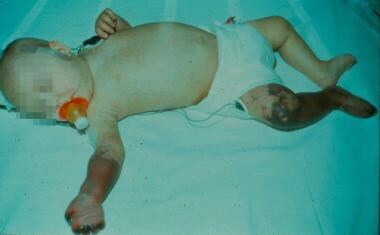 Childhood polyarteritis nodosa. Gangrene of the forearm and lower leg in a 3-month-old infant.
Childhood polyarteritis nodosa. Gangrene of the forearm and lower leg in a 3-month-old infant.
-
Childhood polyarteritis nodosa. Bilateral axillary aneurysms.
-
Childhood polyarteritis nodosa. Gangrene of the forearm and lower leg in a 3-month-old infant.
-
Childhood polyarteritis nodosa. Multiple aneurysms on arteriogram and autopsy specimen.
-
Thrombosed cystic artery in childhood polyarteritis nodosa.
-
Childhood polyarteritis nodosa. Heart with 5 coronary aneurysms.
-
A: Right coronary artery stenosis. B: Stent in place across stenotic segment.
-
Close-up of stent in place.
Tables

What would you like to print?
- Rituximab Not Inferior to Cyclophosphamide in Pediatric Vasculitis
- Vasculitis Patients Need Multiple COVID Vaccine Boosters
- Managing ANCA Vasculitis: Updated British Guideline ‘Transcends’ Specialties
-
 Smartphones and the Future of Psoriasis Care
Smartphones and the Future of Psoriasis Care
-
Joint Pain in Paradise: A Closer Look at Arthritis and HS
-
Immunoglobulin A Nephropathy (IgAN): 5 Differential Diagnoses to Know






