Background
Bernard-Soulier syndrome (BSS) is one of a group of hereditary platelet disorders characterized by thrombocytopenia, giant platelets, and qualitative platelet defects resulting in bleeding tendency. [1, 2, 3] See the image below.
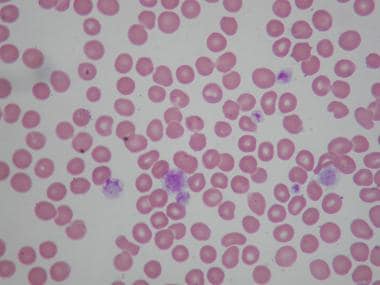 Peripheral smear of patient with Bernard-Soulier syndrome (BSS) showing giant platelets. These platelets are not counted as platelets in most particle counters.
Peripheral smear of patient with Bernard-Soulier syndrome (BSS) showing giant platelets. These platelets are not counted as platelets in most particle counters.
BSS is the second most common inherited platelet defect. Other disorders in the category of macrothrombocytopenia are the May-Hegglin anomaly and gray platelet syndrome.
Treatment is generally supportive. Patients should be educated about the disease and the need to avoid trauma and nonsteroidal anti-inflammatory drugs (NSAIDS). Emphasis should be placed on good dental hygiene.
Pathophysiology and Etiology
The underlying biochemical defect in BSS is the absence or decreased expression of the platelet membrane glycoprotein complex GPIb/IX/V. [4, 5, 6, 7] This complex is the receptor for von Willebrand factor (vWF), and the result of decreased expression is deficient binding of vWF to the platelet membrane at sites of vascular injury, resulting in defective platelet adhesion. The GPIb/IX/V complex is located in the platelet cytoskeleton and hence is also essential to the production of platelets from the megakaryocytes in the bone marrow (which explains the large size and decreased numbers of platelets in this syndrome).
Defective platelet adhesion is demonstrated by the lack of platelet aggregation in response to ristocetin, an antibiotic that normally causes platelets to aggregate. The end result is a lack of formation of the primary platelet plug, together with an increased bleeding tendency. The cause of the thrombocytopenia is not definitely known but is thought probably to be related to the absence of the GPIb/IX/V complex and its role in the production and shedding of platelets from the marrow megakaryocytes. [8]
BSS derives from mutations in GP1BA, GP1BB, and GP9, genes that code for subunits of the GPIb/IX/V complex. Nonsense, missense, and frameshift mutations have been reported. [1] [3]
BSS is inherited in an autosomal recessive fashion; thus, males and females are affected with equal frequency. (A rare variant with autosomal dominant inheritance has been described.) Heterozygotes have been thought to usually have no bleeding manifestations. However, a study by Bragadottir et al indicated that individuals who are heterozygous for BSS mutations have lower platelet counts, more mucocutaneous bleeding, and higher vWF levels than do controls without these mutations, although bleeding is much milder than in persons who are homozygous for BSS. [9]
Epidemiology and Prognosis
BSS is rare, with an estimated occurrence of less than 1 case per million population. Bleeding due to BSS may begin in infancy and may continue with varying severity throughout life, although it may somewhat diminish with age. Males and females are affected with equal frequency. To date, BSS has been described primarily in Whites of European ancestry as well as in the Japanese population; its prevalence in other ethnic groups is unknown.
-
Peripheral smear of patient with Bernard-Soulier syndrome (BSS) showing giant platelets. These platelets are not counted as platelets in most particle counters.
