Background
Elevated blood tyrosine levels are associated with several clinical entities. The term tyrosinemia was first given to a clinical entity based on observations (eg, elevated blood tyrosine levels) that have proven to be common to various disorders, including transient tyrosinemia of the newborn (TTN), hereditary infantile tyrosinemia (tyrosinemia I), Richner-Hanhart syndrome (tyrosinemia II), [1] and tyrosinemia III. In addition, a mysterious entity called tyrosinosis has been described once in the literature. This designation was chosen at a time when specific enzymatic diagnosis was unavailable, leaving a clinical description that has not been duplicated in the 50 years since its publication.
Transient tyrosinemia is believed to result from delayed enzyme maturation in the tyrosine catabolic pathway. This condition is essentially benign and spontaneously disappears with no sequelae. Transient tyrosinemia is not categorized as an inborn error of metabolism because it is not caused by a genetic mutation.
Hereditary infantile tyrosinemia, or tyrosinemia I, is a completely different disease. Patients have a peculiar (cabbagelike) odor, renal tubular dysfunction (Fanconi syndrome), and survival of less than 12 months of life if untreated. Fulminant onset of liver failure occurs in the first few months of life. Some patients have a later onset, usually before age 6 months, with a somewhat protracted course.
For many years, the diagnosis was based on the observation that plasma tyrosine and methionine levels were significantly elevated. Postmortem examination revealed that both the liver and the kidney had a highly unusual pattern of nodular cirrhosis, the histopathologic hallmark of the disease. In the early 1970s, researchers discovered that most severe liver diseases caused such findings regardless of etiology, and, in the late 1970s, the biochemical and enzymatic causes of the disease were reported.
Tyrosinemia II is a disease with a clinical presentation distinctly different from that described above. This presentation includes herpetiform corneal ulcers and hyperkeratotic lesions of the digits, palms, and soles, as well as intellectual disability. The biochemical and enzymatic basis for the disease bears no relationship to that of tyrosinemia I, and tyrosinemia II is not discussed further in this article.
Tyrosinemia III is an extremely rare cause of intermittent ataxia, without hepatorenal involvement or skin lesions, and is also not discussed further in this article.
Pathophysiology
The biochemical basis for tyrosinemia I remained enigmatic until the late 1970s, when researchers described a compound called succinylacetone (SAA) found in the urine of infants with the condition. The normal catabolic pathway, which moves tyrosine to 4-hydroxyphenylpyruvic acid, then to homogentisic acid, the ring then being cleaved to produce maleylacetoacetate (MAA) and fumarylacetoacetate (FAA), is interrupted at the next step, which would normally produce fumarate and acetoacetate. The therapeutic agent 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC) interrupts the pathway after formation of 4-OH-phenylpyruvic acid. Absent this agent, with the accumulation of FAA, there is conversion to succinylacetoacetate, and spontaneous decarboxylation results in production of SAA, which is a metabolic dead-end. See the image below.
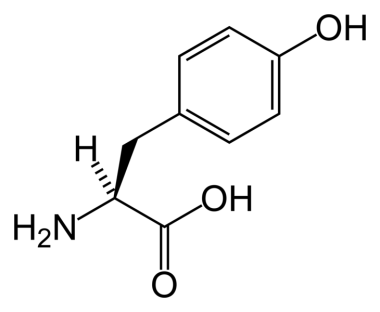 Tyrosine
Tyrosine
Succinylacetone was ultimately determined to be the decarboxylation product of succinyl acetoacetate, a compound derived from the tyrosine catabolic intermediate fumarylacetoacetate. Investigators inferred that the enzymatic defect might reside in deficiency of fumarylacetoacetase, which mediates production of fumaric acid and acetoacetate in both liver and kidney. This inference was later proven correct; succinyl acetoacetate accumulated because of this defect. Decarboxylation produced succinylacetone, which was then excreted in the urine, its source both hepatic and renal.
Although many aspects of the biochemical toxicity of this compound are known, the cellular basis for the multiorgan dysfunction found at the clinical level is unclear. In the kidney, succinylacetone has been demonstrated to be a mitochondrial toxin that inhibits substrate-level phosphorylation by means of the Krebs cycle. This compound also causes dysfunction of membrane transport in normal rat kidneys, altering membrane fluidity and possibly disrupting normal structure. It can cause renal tubular dysfunction in normal rat kidneys, mimicking human Fanconi syndrome, for which no other pharmacological animal model is available. Beyond its effects on the kidney, succinylacetone is a potent inhibitor of δ-aminolevulinic acid dehydratase, the enzyme that mediates formation of porphobilinogen, the cyclic precursor of porphyrins in the heme biosynthetic sequence. Succinylacetone-related alterations in heme biosynthesis of normal rat liver and kidney have been demonstrated.
Data have suggested that fumarylacetoacetate itself induces mitotic abnormalities and instability in the genome. [2] Research in murine animal models has indicated that this metabolite initiates apoptosis of hepatic and renal tubular cells. Taken together, these data form the basis for a unifying hypothesis regarding the development of hepatocellular carcinoma in children with hereditary tyrosinemia. A 2013 report [3] indicates that fumarylacetocetate, but not succinylacetone, inhibits DNA glycosylases, which are instrumental in removing mutagenic base substitutions in the gene. Such data more firmly establish an etiologic relationship between the pathway intermediates and the mutational events they cause.
A new animal model, a rabbit knockout of fumarylacetoacetate hydrolase (FAH), which is the deficient enzyme in the affected human, has been reported; the authors state that the model is a faithful reproduction of the human disease and offers great potential for further study. [4] In recent years, attention has turned toward use of the CRISPR/Cas9 technique to engineer normal cells from affected ones, with an intent to "genetically engineer" affected infants as a replacement for liver transplantation. [5]
The effective therapeutic use of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC) in tyrosinemia does not normalize hepatic collagen metabolism, leaving the already fibrosed liver vulnerable to further structural damage. However, data regarding the hepatic response to NTBC are conflicting.
One group reported reversibility of cirrhotic nodules in a patient receiving NTBC treatment, whereas another group reported that the drug did little to suppress gene expression of other genes responsible for ongoing hepatic damage in a murine model. Longer-term experience with NTBC has provided more encouraging results, suggesting that children with the disease who receive NTBC prior to age 2 years have less than a 5% risk of developing hepatocarcinoma.
A 2014 report indicated rapid reversal with NTBC treatment (within 48 hours) of phosphaturia and continuing improvement in other parameters of renal tubular dysfunction over the ensuing 2 weeks of the study. [6] In addition, corneal opacities due to deposition of tyrosine crystals in the tissues caused by long-term treatment with NTBC have been reported. Tyrosine levels rise because of the enzymatic block created in the p -hydroxyphenylpyruvic acid dioxygenase. A tendency for precipitation is noted because tyrosine is relatively insoluble compared with other amino acids.
The potential long-term effects of precipitation in tissues other than the cornea remain unknown, although data so far indicate there are no ill effects. [7] On the other hand, long-term neurocognition studies indicate that treated children with a frequently elevated serum tyrosine significantly lag behind their siblings. [8, 9, 10, 11]
In patients with tyrosinemia who have undergone orthoptic liver transplantation, urinary excretion of succinylacetone dramatically decreases, although excretion generally persists at levels lower than those observed before transplantation. [12] This persistence can be attributed to ongoing production of the compound by kidneys, which remain genetically affected by the enzyme defect. The generalized toxic effect on mitochondria, membranes, and heme biosynthesis can logically be assumed to be at the root of the pathologic observations of nodular cirrhosis.
Increased urinary excretion of δ-aminolevulinic acid can be attributed to inhibition of the heme biosynthetic pathway. A similar mechanism can account for the seizures commonly observed in patients; this mechanism is based on the demonstration of fumarylacetoacetase in the normal human brain. Absence of normal enzyme function could then be assumed to induce cellular accumulation of succinylacetone and to facilitate its toxic effects on the neuron.
Epidemiology
Frequency
United States
The estimated incidence is 1 case per 100,000 live births.
International
In some areas of North America, notably a region of Quebec province, the incidence is extraordinarily high, and the estimated incidence of carriers of a specific mutation is 1 in 14 adults.
Mortality/Morbidity
Affected infants often have a fulminant onset, with a rapid development of hepatic cirrhosis and failure. The onset of hepatic failure places the infant at risk for a serious coagulopathy. Survivors of the neonatal episode are at significant risk of hepatocellular carcinoma. In one series in which combined medical and surgical techniques were used, the mortality rate was reduced to less than 15%.
Sex
Tyrosinemia I is an autosomal recessive disorder; therefore, the sex distribution is equal. The severity of onset and the subsequent course does not differ between the sexes. To date, 95 causative mutations have been described. [13]
Age
The disease is present from conception because it is caused by genetic mutation. Most infants present within the first 2-3 months of life; far fewer infants present later with a chronic form, which frequently manifests initially as rickets and slowly developing hepatic cirrhosis.
Prognosis
In all initially diagnosed cases, including those discovered in early infancy, prognosis should be regarded as guarded. However, in the very young infant, early institution of NTBC treatment promises a far better eventual outcome than in those patients treated later. As with most inherited metabolic disorders with long-term survival, it is becoming apparent that appropriate maintenance of therapy becomes very difficult in the adolescent period. [14]
-
Tyrosine




