Practice Essentials
Wolf-Hirschhorn syndrome (WHS), also known as deletion 4p syndrome, is a disorder caused by irregularities on the short arm of chromosome 4 (with these irregularities signified as 4p-). It is characterized by a distinctive facial appearance (the Greek warrior helmet), growth delay, intellectual disability, hypotonia, and seizures.
Cooper and Hirschhorn first documented WHS in 1961. [1] They described a child with midline fusion defects. In 1965, back-to-back publications in Humangenetik by Hirschhorn et al and Wolf et al brought the disease to the attention of geneticists and other medical professionals. [2, 3] Numerous cases were subsequently published. (See the images below.)
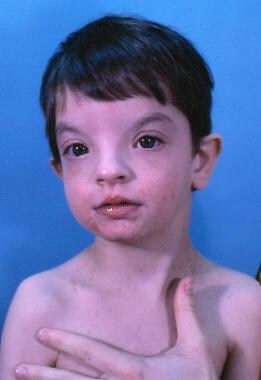 A child with Wolf-Hirschhorn syndrome. Note the characteristic dysmorphic facial features, including prominent glabella, hypertelorism, beaked nose, and frontal bossing, collectively described as "Greek warrior helmet" facies.
A child with Wolf-Hirschhorn syndrome. Note the characteristic dysmorphic facial features, including prominent glabella, hypertelorism, beaked nose, and frontal bossing, collectively described as "Greek warrior helmet" facies.
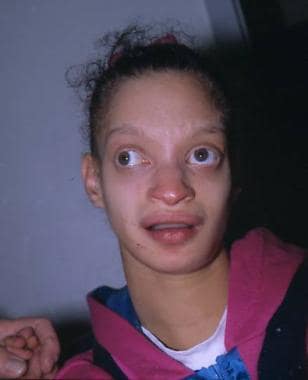 A girl with Wolf-Hirschhorn syndrome showing characteristic features of the condition.
A girl with Wolf-Hirschhorn syndrome showing characteristic features of the condition.
Signs and symptoms of Wolf-Hirschhorn syndrome
The following findings are present in greater than 75% of patients [4, 5] :
-
Typical facial appearance - "Greek warrior helmet" appearance of the nose (wide nasal bridge continuing to the forehead), high-arched eyebrows, hypertelorism, epicanthus, high anterior hairline, short philtrum, downturned corner of the mouth
-
Microcephaly
-
Low-set ears with pits or tags
-
Intrauterine growth restriction (IUGR)/postnatal growth deficiency
-
Hypotonia with decreased muscle bulk
The following findings are present in 50-75% of patients [4, 5] :
-
Skeletal anomalies - Kyphosis and scoliosis with malformed vertebral bodies, accessory or fused ribs, clubfeet, split hand
-
Skin - Hemangiomas, marble and/or dry skin
-
Craniofacial asymmetry
-
Dental anomalies - Delayed dentition, taurodontism in the primary dentition, peg-shaped teeth, dental agenesis
-
Ptosis
The following findings are present in 25-50% of patients [4, 5] :
-
Heart murmur secondary to congenital heart defect, most commonly atrial septal defects; pulmonary stenosis, ventricular septal defects, patent ductus arteriosus, aortic insufficiency, and tetralogy of Fallot are also seen
-
Ophthalmologic anomalies - Exodeviation, nasolacrimal obstruction, eye or optic nerve coloboma
-
Unilateral or bilateral cleft lip/palate
-
Stereotypies - Hand washing/flapping, rocking
Of note, central nervous system anomalies are present in one third of cases, and seizures occur in 50-100% of patients.
Workup in Wolf-Hirschhorn syndrome
Diagnosis is made by confirming a deletion in the WHS critical region (WHSCR) using conventional chromosome analysis, fluorescence in situ hybridization (FISH), or chromosomal microarray.
Imaging studies in WHS include the following:
-
Radiography - May reveal delayed bone maturation, microcephaly, hypertelorism, micrognathia, anterior fusion of vertebrae, fused ribs, dislocated hips, proximal radioulnar synostosis, clubfeet, and split hand/foot abnormality
-
Echocardiography - Should be obtained in all patients to evaluate for heart defects
-
Renal ultrasonography - Used to detect renal anomalies
-
Magnetic resonance imaging (MRI) and computed tomography (CT) scanning - May reveal underlying brain pathology, including agenesis of the corpus callosum and ventriculomegaly
Management of Wolf-Hirschhorn syndrome
Medical care is supportive in patients with WHS. The underlying disorder has no known treatment. Management can include the following:
-
Multidisciplinary team approach, including speech and communication therapy and sign language
-
Standard care for skeletal anomalies, ophthalmologic abnormalities, congenital heart defects, and hearing loss
-
Genetic counseling
-
Gavage feeding and/or gastrostomy for feeding difficulties
-
Seizure control - Sodium bromide is a helpful drug for many patients with WHS, particularly to prevent status epilepticus; [6] valproic acid is used for atypical absence seizures, and benzodiazepines are also indicated in status epilepticus
Pathophysiology
WHS results from irregularities of the distal short arm of chromosome 4, including deletions of varying size (most common) and microduplications. Several genetic mechanisms have been described (see the image below):
-
De novo deletion (50-60% of cases)
-
Unbalanced translocation (40-45% of cases) - Either de novo or inherited from a parent with a balanced translocation
-
Other complex rearrangements, leading to a 4p16.3 deletion - Ring 4 chromosome, 4p-mosaicism
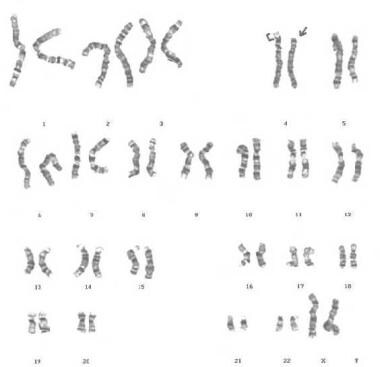 G-banded karyotype showing deletion of 4p, derived from the mother, with balanced translocation (4p;8p).
G-banded karyotype showing deletion of 4p, derived from the mother, with balanced translocation (4p;8p).
Diagnosis is made by confirming a deletion in the WHS critical region (WHSCR) using conventional chromosome analysis, fluorescence in situ hybridization (FISH), or chromosomal microarray.
Evidence strongly supports the idea that the ‘‘core’’ phenotype (distinctive facial appearance, intellectual disability, growth delay, and seizures or electroencephalographic anomalies) is due to haploinsufficiency of several genes on distal 4p, including WHSC1, WHSC2, TACC3, FGFR3, and LETM1. [7, 8] Therefore, WHS represents a contiguous gene syndrome with contribution of genes located in the terminal 4p16.3 region, within a 1.5-1.6 Mb region.
The presence of a true genotype-phenotype correlation is controversial, as some studies have indicated that there is not always a correlation between the deletion's size and the severity of the clinical findings [4] .
Epidemiology
Frequency
United States
The frequency of WHS is estimated to be 1 case per 50,000 births to 1 case per 20,000 births. [9, 10]
Mortality/Morbidity
The mortality rate is estimated at 34% in the first 2 years of life. However, because many affected children die before the diagnosis is confirmed or suspected, the mortality rate is underestimated. The usual cause of death is a heart defect, aspiration pneumonia, infection, or seizure.
The prenatal mortality rate of Wolf-Hirschhorn syndrome is not significantly increased because 4p deletions are not reported in spontaneous abortions.
Associated adulthood morbidity includes sequelae from congenital heart defects; marked growth failure; contracture of hands, wrists, and feet; poor development of secondary sexual characteristics; and severe growth and intellectual impairment.
Race
Wolf-Hirschhorn syndrome has no ethnic predilection.
Sex
Wolf-Hirschhorn syndrome is more common in females than in males, with a male-to-female ratio of 1:2.
Age
Usually, the condition is detected in the newborn period because of dysmorphic features.
Prognosis
Patients with WHS have many health-associated difficulties, particularly in the first 3 to 5 years, with these problems stemming primarily from growth failure and difficult-to-treat seizures. Neurodevelopmental skills are noted to progress slowly but constantly. In the absence of major malformations, patients with WHS have a life expectancy similar to that of other patients with a congenital seizure disorder and developmental disability.
Patients with WHS are at risk for the following:
-
Seizures
-
Failure to thrive
-
Cognitive deficits
-
Motor deficits
-
Deficits in autonomy and sphincter control
Seizures
Seizures are a major concern for patients with WHS and are typically hard to control, especially early on. However, rapid diagnosis and treatment leads to improved control and outcomes. The majority of patients with WHS experience a decrease in the frequency of seizures after age 5 years, [6] and in approximately 50% of patients, seizures resolve by age 13 years.
Failure to thrive
Growth parameters are typically below the second percentile. Growth charts specific for children with WHS are available. [11]
Cognitive deficits
Severe intellectual disabilities are observed in the majority of patients (65%); however a broad range can be seen, including mild and moderate intellectual disability in 10% and 25% of patients, respectively. [12]
Expressive language is typically limited to guttural or disyllabic sounds. However, a minority of patients (< 10%) are able to communicate using simple sentences.
Comprehension is typically context specific and limited to simple orders. Intent to communicate is present in most patients.
Motor deficits
About 45% of patients are able to walk, either independently (25%) or with support (20%). Walking is significantly delayed in these patients, being achieved between ages 2 and 12 years.
Autonomy and sphincter control
Only about 10% of patients achieve daytime sphincter control (typically between ages 8 and 14 years). Ten percent of patients are able to feed themselves, while about 20% can assist with dressing/undressing.
Adult health risks
In adulthood, patients with WHS are at risk for the following:
-
Chronic kidney disease
-
Malignancies - Specifically, liver adenomas
Patient Education
The following websites contain updated information for patients and their families:
In addition, up-to-date information about the syndrome should be made available to families through the following organizations:
-
A child with Wolf-Hirschhorn syndrome. Note the characteristic dysmorphic facial features, including prominent glabella, hypertelorism, beaked nose, and frontal bossing, collectively described as "Greek warrior helmet" facies.
-
A fetus with Wolf-Hirschhorn syndrome. Note the presence of "Greek warrior helmet" facies.
-
The result of a fluorescence in situ hybridization (FISH) study of a patient with Wolf-Hirschhorn syndrome. FISH photograph shows deletion of a locus-specific probe for the Wolf-Hirschhorn critical region (absence of a probe signal at 4p16.3).
-
G-banded karyotype showing deletion of 4p, derived from the mother, with balanced translocation (4p;8p).
-
A girl with Wolf-Hirschhorn syndrome showing characteristic features of the condition.
