Practice Essentials
Down syndrome is by far the most common and best known chromosomal disorder in humans and the most common cause of intellectual disability. It primarily results from trisomy of chromosome 21 (see the image below), which gives rise to multiple systemic complications as part of the syndrome. Levels of ability and challenges range widely in individuals with Down syndrome. Some need significant support throughout life, while others grow up to hold jobs and live on their own.
Clinical features of Down syndrome
Individuals with Down syndrome typically exhibit distinct dysmorphic features, primarily affecting the head, neck, and extremities. Cardiovascular anomalies, particularly congenital heart defects (such as septal defects), are present in about half of those with Down syndrome, and gastrointestinal issues such as duodenal atresia and esophageal abnormalities are also prevalent.
Infants with Down syndrome often have slower growth, lower birth measurements, and feeding difficulties. Delayed milestones are universal, and persons with Down syndrome are at high risk for obesity in early childhood. Ophthalmic and hearing problems are frequent, including cataracts and hearing loss, and endocrine issues like thyroid dysfunction are common. Neuropsychiatric concerns are common as well, with most individuals experiencing cognitive impairment and some displaying behavioral or psychiatric conditions. Many females with Down syndrome are fertile, while most males are not.
Additional health challenges include atlantoaxial instability, hematologic abnormalities (eg, polycythemia, leukemia), immunologic impairments, and increased susceptibility to various infections, urologic conditions, and joint and skin disorders.
See Presentation for more detail.
Diagnosis of Down syndrome
Down syndrome is often diagnosed prenatally; if no prenatal diagnosis is made, the syndrome can typically be recognized by its distinct physical features. Confirmation requires genetic testing, usually a karyotype on a blood sample, to identify trisomy 21 or other chromosomal variants like translocations or mosaicism.
See Workup for more detail.
Management
Down syndrome management involves multidisciplinary care focused on early intervention, regular health screenings, and supportive therapies. Key aspects include monitoring for heart, thyroid, vision, and hearing issues, along with developmental support through physical, occupational, and speech therapies. Regular specialist care and access to family and community resources are essential to enhance quality of life and development.
There are no medical treatments for intellectual disability associated with Down syndrome, but improved medical care has greatly enhanced the quality of life for individuals with this syndrome and has increased their life expectancy.
See Treatment and Medication for more detail.
Background
As previously stated, Down syndrome is by far the most common and best known chromosomal disorder in humans and the most common cause of intellectual disability. [1, 2, 3, 4, 5] In addition to intellectual disability, it is characterized by dysmorphic facial features and other distinctive phenotypic traits. Down syndrome is caused primarily by trisomy of chromosome 21; this is the most common trisomy among live births. The term mongolism was once commonly used for Down syndrome but is now considered obsolete. [6, 7, 8]
Like most diseases associated with chromosomal abnormalities, trisomy 21 gives rise to multiple systemic complications as part of the clinical syndrome. This chromosomal anomaly leads to both structural and functional defects in patients with Down syndrome. However, not all defects occur in each patient; there is a wide range of phenotypic variation. [9]
Pathophysiology
The extra chromosome 21 affects almost every organ system and results in a wide spectrum of phenotypic consequences. These include life-threatening complications, clinically significant alteration of life course (eg, intellectual disability), and dysmorphic physical features. Down syndrome decreases prenatal viability and increases prenatal and postnatal morbidity. Affected children have delays in physical growth, maturation, bone development, and dental eruption.
Two different hypotheses have been proposed to explain the mechanism of gene action in Down syndrome: developmental instability (ie, loss of chromosomal balance) and the so-called gene-dosage effect. [10] According to the gene-dosage effect hypothesis, the genes located on chromosome 21 are overexpressed in cells and tissues of Down syndrome patients, and this contributes to the phenotypic abnormalities. [11]
Containing about 33 genes, the Down syndrome critical region (DSCR), on chromosome 21 at 21q22.1-q22.3, plays a pivotal role in the pathophysiology of Down syndrome. Its significance was identified through studies of individuals with partial trisomies of chromosome 21, which linked the presence of extra copies of the DSCR to typical Down syndrome features. [12]
Within the DSCR, genes like DYRK1A and DSCR1/RCAN1 impact cognitive function and cardiac signaling, leading to intellectual disabilities and congenital heart defects. DYRK1A is also implicated in causing the characteristic facial dysmorphisms seen in Down syndrome, such as a flattened facial profile and slanted eyes. Outside the DSCR, genes like APP, influencing Alzheimer-like cognitive decline; CBS, affecting cardiovascular health; and SOD1, linked to oxidative stress and premature aging, further contribute to the Down syndrome phenotype.
Abnormal physiologic functioning affects thyroid metabolism and intestinal malabsorption. Patients with trisomy 21 have an increased risk of obesity. Frequent infections are presumably due to impaired immune responses, and the incidence of autoimmunity, including as seen in the development of hypothyroidism and rare Hashimoto thyroiditis, is increased.
Patients with Down syndrome have decreased buffering of physiologic reactions, resulting in hypersensitivity to pilocarpine and abnormal responses on sensory-evoked electroencephalographic (EEG) tracings. Children with leukemic Down syndrome also have hyperreactivity to methotrexate.
Decreased buffering of metabolic processes results in a predisposition to hyperuricemia and increased insulin resistance. Diabetes mellitus develops in many affected patients. Premature senescence causes cataracts and Alzheimer disease.
Musculoskeletal manifestations in patients with Down syndrome include reduced height, atlanto-occipital and atlantoaxial hypermobility, and vertebral malformations of the cervical spine. These findings may lead to atlanto-occipital and cervical instability as well as complications such as weakness and paralysis.
A study by Romano et al indicated that in persons with Down syndrome, brain cortical thickness is reduced with increasing age. The study involved 91 persons with Down syndrome, none of whom had dementia, with cortical thickness measured using magnetic resonance imaging (MRI). Frontal, temporal, parietal, and cingulate gyrus measurements showed bilateral cortical thinning in association with age, with thickness apparently declining more significantly and rapidly between the ages of 20 and 30 years. [13]
Etiology
Down syndrome is caused by the following three cytogenic variants:
-
Three full copies of chromosome 21
-
Chromosomal translocation that results in three copies of the DSCR
-
Mosaicism
In 94% of patients with Down syndrome, full trisomy 21 is the cause; mosaicism (2.4%) and translocations (3.3%) account for the remaining cases. Approximately 75% of the unbalanced translocations are de novo, and approximately 25% result from familial translocation.
A free trisomy 21 results from nondisjunction during meiosis in one of the parents. This occurrence is correlated with advanced maternal and paternal age. About 95% of the time, the error is maternal nondisjunction, with meiosis I errors occurring three times as frequently as meiosis II errors. The remaining 5% cases are paternal in origin, and meiosis II errors predominate.
Advanced maternal age remains the only well-documented risk factor for maternal meiotic nondisjunction. Research has uncovered several age-related changes in oocytes that elevate the risk for nondisjunction, contributing to trisomy 21. The main factors identified are cohesin protein degradation, weakened spindle assembly checkpoints, mitochondrial dysfunction with oxidative stress, and recombination errors. [14, 15]
Translocation occurs when genetic material from chromosome 21 becomes attached to another chromosome, resulting in 46 chromosomes, with 1 chromosome having extra material from chromosome 21 attached. It may occur de novo or be transmitted by one of the parents. Translocations are usually of the centric fusion type. They frequently involve chromosome 14 (14/21 translocation), chromosome 21 (21/21 translocation), or chromosome 22 (22/21 translocation).
Mosaicism is considered a postzygotic event (ie, one that occurs after fertilization). Most cases result from a trisomic zygote with mitotic loss of one chromosome. As a result, two cell lines are found: one with a free trisomy and the other with a normal karyotype. This finding leads to great phenotypic variability, ranging from a near normal phenotype to the classic trisomy 21 phenotype.
Cytogenetic and molecular studies suggest that dup21(q22.1-22.2) is sufficient to cause Down syndrome. The DSCR contains genes that code for enzymes, such as SOD1, CBS, and GARS-AIRS-GART.
Epidemiology
Down syndrome is the most common autosomal abnormality. Approximately 1 in 700 newborns in the United States has Down syndrome, or about 5300 babies annually. [16] Down syndrome accounts for about one third of all moderate and severe mental handicaps in school-aged children. The prevalence of Down syndrome worldwide has increased because of increases in life span.
Maternal age and diagnosis of Down syndrome
Down syndrome can be diagnosed prenatally with amniocentesis, percutaneous umbilical blood sampling (PUBS), chorionic villus sampling (CVS), and extraction of fetal cells from the maternal circulation. It is often diagnosed shortly after birth by recognizing dysmorphic features and the distinctive phenotype. The characteristic morphologic features will be obvious in children older than 1 year. Some dermatologic features increase with advancing age.
Occurrence is strongly dependent on maternal age. The incidence of this syndrome at various maternal ages is as follows:
-
15-29 years - 1 case in 1500 live births
-
30-34 years - 1 case in 800 live births
-
35-39 years - 1 case in 270 live births
-
40-44 years - 1 case in 100 live births
-
Older than 45 years - 1 case in 50 live births
On rare occasions, the disease can be observed in a few members of a family. The risk for recurrence of Down syndrome in a patient’s siblings also depends on maternal age.
Sex-related demographics
Overall, the two sexes are affected roughly equally. The male-to-female ratio is slightly higher (approximately 1.15:1) in newborns with Down syndrome, but this effect is restricted to neonates with free trisomy 21.
Thirty-five percent to 50% of female patients with trisomy 21 are fertile, and these females have up to a 50% chance of having a live child who also has trisomy 21. (However, many affected fetuses abort spontaneously.) On the other hand, men with Down syndrome are usually infertile, except for those with mosaicism.
Race-related demographics
Down syndrome has been reported in people of all races; no racial predilection is known. African American patients with Down syndrome have substantially shorter life spans than White patients with trisomy 21.
Prognosis
The overall outlook for individuals with Down syndrome has dramatically improved. Many adult patients are healthier and better integrated into society, and life expectancy has risen from 25 years in 1983 to approximately 60 years today. [17]
About 75% of concepti with trisomy 21 die in embryonic or fetal life. Approximately 25-30% of patients with Down syndrome die during the first year of life. The most frequent causes of death are respiratory infections (bronchopneumonia) and congenital heart disease.
In addition, esophageal atresia with or without transesophageal (TE) fistula, Hirschsprung disease, duodenal atresia, and leukemia contribute to mortality. The high mortality later in life may be the result of premature aging.
In elderly persons with Down syndrome, relative preservation of cognitive and functional ability is associated with better survival. [18] Clinically, the most important disorders related to mortality in this population are dementia, mobility restrictions, visual impairment, and epilepsy (but not cardiovascular disease). In addition, the level of intellectual disability and institutionalization are associated with mortality.
Individuals with Down syndrome have a greatly increased morbidity, primarily because of infections involving impaired immune response. Large tonsils and adenoids, lingual tonsils, choanal stenosis, or glossoptosis can obstruct the upper airway. Airway obstruction can cause serous otitis media, alveolar hypoventilation, arterial hypoxemia, cerebral hypoxia, and pulmonary arterial hypertension with resulting cor pulmonale and heart failure.
Leukemia, thyroid diseases, autoimmune disorders, epilepsy, intestinal obstruction, and increased susceptibility to infections (including recurrent respiratory infections) are commonly associated with Down syndrome.
The aging process seems to be accelerated in patients with Down syndrome. The Alzheimer disease–associated neuropathologic characteristics of amyloid-β plaques and neurofibrillary tangles made of hyperphosphorylated tau are found in almost all persons with Down syndrome by age 40 years. [17] Alzheimer disease itself develops in about 50% of individuals with Down syndrome, often arising at a relative early age. By the seventh decade of life, persons with Down syndrome have a more than 90% chance of developing Alzheimer disease. [17]
A delay in recognizing atlantoaxial and atlanto-occipital instability may result in irreversible spinal-cord damage. Visual and hearing impairments in addition to intellectual disability may further limit the child’s overall function and may prevent him or her from participating in important learning processes and developing appropriate language and interpersonal skills. Unrecognized thyroid dysfunction may further compromise central nervous system (CNS) function.
A questionnaire study by Matthews et al of caregivers of persons with Down syndrome aged 20 years or older reported that, while adults with Down syndrome who had a greater amount of health issues tended not to be independent and social and although current health problems impacted communication skills in these individuals, the number of congenital abnormalities in adults with Down syndrome was not significantly associated with scores for independence/life skills. [19]
Patient Education
Career preparation should include acquisition of job skills, choice of job area, development of work-support behavior, and opportunities for job mobility. The goal of successful transition from school to a work situation is meaningful employment and optimal function in the least restrictive environment.
Opportunities to participate in community life should be made available. Individuals should be encouraged to pursue daily living tasks with minimal or no assistance. They should participate in cultural, leisure, and recreational activities during the growing years. Patients may qualify for supplemental security income (SSI), depending on their family’s income.
Parents may benefit from joining a local Down syndrome support group. Additional resources can be obtained from the following organizations:
-
Infant with Down syndrome. Note up-slanting palpebral fissures, bilateral epicanthal folds, flat nasal bridge, open mouth with tendency for tongue protrusion, and small ear with overfolded helix.
-
Child with Down syndrome. Note up-slanting palpebral fissures, bilateral epicanthal folds, small nose with flat nasal bridge, open mouth with tendency for tongue protrusion, and small ears with overfolded helix.
-
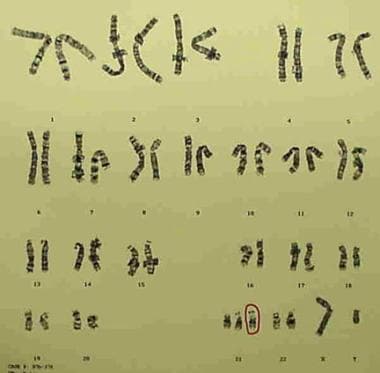
G-banded karyotype showing trisomy 21 (47,XY,+21).
-
G-banded karyotype showing trisomy 21 of isochromosome arm 21q type [46,XY,i(21)(q10)].
-
Hand of infant with Down syndrome. Note transverse palmar crease and clinodactyly of fifth finger.
-
Ear of infant with Down syndrome. Note characteristic small ear with overfolded helix.
-
Characteristic flat facies with hypertelorism, depressed nasal bridge, and protrusion of tongue, as well as single palmar simian crease, in girl aged 2 years with Down syndrome. Image courtesy of L. Dourmishev, MD, PhD, DSc.
-
Small auricle and anomalies of folds in patient with Down syndrome. Image courtesy of L. Dourmishev, MD, PhD, DSc.
-
Palmar simian crease in patient with Down syndrome. Image courtesy of L. Dourmishev, MD, PhD, DSc.
-
Patient with Down syndrome with protuberant abdomen and umbilical hernia. Image courtesy of L. Dourmishev, MD, PhD, DSc.
-
Wide gap between first and second toes and onychomycosis in patient with Down syndrome. Image courtesy of L. Dourmishev, MD, PhD, DSc.
-
Hypodontia in patient with Down syndrome. Image courtesy of L. Dourmishev, MD, PhD, DSc.
