Background
Double outlet right ventricle (DORV), as depicted in the image below, is a type of ventriculoarterial connection in which both the aorta (AO) and pulmonary artery (PA) arise entirely or predominantly from the right ventricle (RV). The only outlet from the left ventricle (LV) is a ventricular septal defect (VSD).
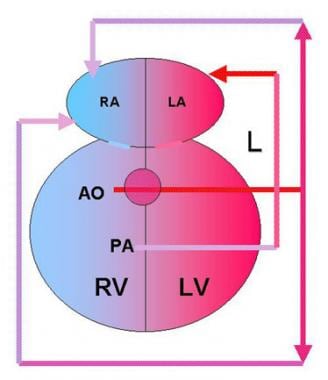 Double outlet right ventricle (DORV) with transposition of the great arteries accounts for 26% of cases of DORV. The aorta (AO) is anterior and to the right of the pulmonary artery (PA), and both arteries arise from the right ventricle (RV). The only outflow from the left ventricle (LV) is a ventricular septal defect (VSD), which diverts blood toward the RV. Pulmonary veins drain into the left atrium (LA) after blood has been oxygenated in the lungs (L). Systemic venous return is to the right atrium (RA).
Double outlet right ventricle (DORV) with transposition of the great arteries accounts for 26% of cases of DORV. The aorta (AO) is anterior and to the right of the pulmonary artery (PA), and both arteries arise from the right ventricle (RV). The only outflow from the left ventricle (LV) is a ventricular septal defect (VSD), which diverts blood toward the RV. Pulmonary veins drain into the left atrium (LA) after blood has been oxygenated in the lungs (L). Systemic venous return is to the right atrium (RA).
DORV is usually associated with concordant atrioventricular (AV) connections (ie, the right atrium drains into the RV and the left atrium drains into the LV). Fibrous discontinuity is present between the mitral and semilunar valves. Conus is present beneath both the aortic and pulmonary valves (subpulmonic and subaortic conus).
DORV is virtually always associated with a VSD and, occasionally, with an atrial septal defectPatients with DORV may also present with varying degrees of left ventricular hypoplasia and mitral valve anomalies such as stenosis or atresia. Straddling of the AV valves across the VSD may be present. The aortic valve may be stenosed, and the aortic arch may show coarctation or even interruption. Anomalies of the coronary arteries (CAs), such as those that occur in patients with dextro-transposition of the great arteries may be present. These include the left circumflex arising from the right main, a single right CA, a single left CA, and inverted origin of the CA.
Pathophysiology
The pathophysiology of DORV varies, irrespective of the great arterial relationship (ie, side-by-side, d-transposition of the great arteries, l-transposition of the great arteries, normally related). Clinical manifestations may range from that of a large VSD to that of transposition of the great arteries and may mostly depend on the position of the VSD in relation to the great vessels (whether it is subpulmonary or subaortic) and the presence or absence of pulmonary valve stenosis (PS). Both of these factors contribute substantially to the hemodynamics of this congenital heart defect.
In cases of a subaortic VSD, which occurs in 60-70% of patients, the VSD is closer to the aortic valve, thus oxygenated blood from the LV is directed to the AO and desaturated blood from the right ventricle (RV) is directed primarily to the PA (see the image below).
Double outlet right ventricle (DORV) with transposition of the great arteries accounts for 26% of cases of DORV. The aorta (AO) is anterior and to the right of the pulmonary artery (PA), and both arteries arise from the right ventricle (RV). The only outflow from the left ventricle (LV) is a ventricular septal defect (VSD), which diverts blood toward the RV. Pulmonary veins drain into the left atrium (LA) after blood has been oxygenated in the lungs (L). Systemic venous return is to the right atrium (RA).
PS occurs commonly which forces some desaturated blood into the AO. Because of the large VSD, the RV and the LV as well as the AO handle equal systolic pressures. When PS is present, this poses a restriction to flow to the pulmonary circuit, and thus, systolic pressure in the pulmonary arteries is lower. This physiology resembles that of TOF with cyanosis and no congestive heart failure (CHF).
In cases of a subaortic VSD with no PS, systolic pressure in both great vessels as well as in both ventricles is equal; thus, blood follows the path of least resistance (ie, usually towards the lungs) and the clinical picture is that of a large VSD. The degree of blood oxygenation in the systemic as well as the pulmonary circuits is determined by degree of mixing in the systemic (ie, right) ventricle, which, in turn, depends on the degree of resistance upstream of the pulmonary valve.
All patients with increased pulmonary blood flow (PBF) at systemic or near systemic pressures are at increased risk of developing early pulmonary obstructive vascular disease regardless of their arterial oxygen saturation (ie, presence or absence of cyanosis).
With a subpulmonary VSD (Taussig-Bing anomaly), which occurs in 10% of patients, oxygenated blood from the LV is directed to the PA and desaturated blood from the RV is directed to the AO. This physiology resembles transposition of the great arteries with a VSD; thus, the patient presents with both cyanosis and CHF.
In cases of a doubly committed VSD, the left ventricular outflow is not committed preferentially to either semilunar valve. In the presence of PS, the physiology resembles that of TOF, and in the absence of PS, it is that of a large VSD.
In cases with remote VSD, the VSD is far from both semilunar valves. It is most commonly an AV canal-type VSD. Again, the physiology is that of TOF in cases involving PS and is that of a large VSD when flow through the pulmonary valve is not restricted (ie, absence of PS).
Etiology
DORV is thought to be the result of a malformation in the outlet portion of the embryonic ventricular loop at 3-4 weeks' gestation. Although mostly sporadic, familial cases have been reported.
Fluorescence in situ hybridization (FISH) analysis has shown deletions in the 22q11.2 region in certain individuals with TOF, DORV, transposition of the great arteries, and VSD associated with other congenital heart disease (CHD). [1, 2, 3] As a matter of fact, DORV may be part of complex CHD in patients with DiGeorge syndrome, velocardiofacial syndrome, and conotruncal anomaly–face syndrome.
DORV has also been associated with trisomies 13 and 18 and tetrasomy 8p.
DORV has also been reported in patients with mutations in human cardiac transcription factor NKX2.5.
DORV, truncus arteriosus (TA), atrial septal defect (ASD), atrioventricular septal defect (AVSD), ventricular septal defect (VSD), transposition of the great arteries (TGA), and tetralogy of Fallot (TOF) occur with a higher incidence in the offspring of mothers with pregestational diabetes mellitus than in the general population. Teratogenic mechanisms involved are thought to be related to increased reactive oxygen species production, impaired cell proliferation, and altered Gata-4, Gata-5, and vascular endothelial growth factor (VEGF)–α expression. According to research studies in pregnant diabetic rats, antioxidant supplementation with vitamin E reduced the severity of malformations in their offspring [4] and supplementation of their drinking water with N -acetylcysteine eliminated the incidence of AVSD, TOF, and TGA and decreased the incidence of ASD and VSD. [5]
DORV has been reported to occur in mouse embryos homozygous for the JMJ mutation, which affects the nuclear protein jmj coded by chamber-specific genes.
Studies using animal models described a transcription factor that plays a critical role in directing cardiac asymmetric morphogenesis, known as Pitx2. Specifically, ectopic Pitx2c expression in the developing myocardium was found to correlate with the development of DORV. Whereas loss of function of the Pitx2 caused atrial isomerism, double inlet left ventricle, transposition of the great arteries, persistent truncus arteriosus, and abnormal aortic arch remodeling.
Most recently, hearts with persistent truncus arteriosus, DORV, and transposition of the great arteries, have been postulated to have rotation of the myocardial wall of the outflow tract that is arrested or fails to initiate. This is supported by the discovery that mutations in the NPHP4 gene involved in the formation of motile cilia in the Kupffer vesicle, which produce asymmetrical fluid flow necessary for normal left-to-right asymmetry, cause laterality defects such as dextrocardia, transposition of great arteries, DORV, and caval vein abnormalities. [6]
In summary, the pathogenesis of DORV is currently believed to include impairment of neural crest–derivative migration and impairment of normal cardiac situs and looping. [7]
Epidemiology
United States data
Congenital heart disease (CHD) occurs in less than 1% of all newborns, and DORV is present in 0.5-1.5% of all patients with CHD. The estimated frequency of DORV is 1 case per 10,000 live births. A recent study showed a higher prevalence of DORV, tetralogy of Fallot, and truncus arteriosus, in addition to endocardial cushion defects and single ventricle, in certain regions of the country. [8]
Sex-related demographics
No sex predilection is reported.
Age-related demographics
The presentation is usually in the newborn period with this entity; however, in some circumstances (eg, subaortic VSD with mild-to-moderate PS), the diagnosis may not be made until later in infancy.
Prognosis
Improvement in surgical techniques in recent years has resulted in good outcomes for most patients born with congenital heart disease (CHD). Prognosis for infants born with DORV and transposition of the great arteries is generally good, although it is dependent on specific anatomy. For example, patients with DORV and transposition of the great arteries with a subaortic VSD and no other anatomic abnormalities (eg, left ventricular hypoplasia) are likely to do well after surgery. Patients with restrictive VSD may not do as well because this is a particularly difficult problem. [9] Enlargement of VSD is difficult and likely to result in complications, such as conduction abnormalities (atrioventricular [AV] block).
Morbidity/Mortality
Morbidity and mortality depend not only on the overall clinical condition of the patient at the time of presentation, but also on the type and severity of associated anomalies.
Irrespective of the great vessel relationship, the mortality rate is less than 5% for DORV cases with subaortic VSD and is somewhat higher for those with a doubly committed VSD.
In cases of subpulmonary VSD (Taussig-Bing anomaly), morbidity and mortality depend on whether the patient has already developed pulmonary vascular obstructive disease and also on the type of surgery that is required. [10] In cases of DORV with d-transposition of the great arteries, creation of an intraventricular tunnel between the VSD and the AO carries a mortality risk of 10-15%. In subpulmonary VSD with PS (ie, TOF-type physiology), an intraventricular tunnel between the VSD and the AO in addition to relief of PS by a patch graft also carries a mortality risk of 10-15%. In cases of remote VSD, the preferred surgical repair is creation of an interventricular tunnel between the VSD and the AO. However, it carries a mortality rate as high as 30-40%.
When the above surgical procedures cannot be performed (ie, hypoplastic LV, inadequate anatomy for an intracardiac conduit between the LV and the AO, hypoplastic AO, hypoplastic/atretic mitral valve), a Fontan-type operation is the choice; the mortality rate has decreased to approximately 5%.
-
Double outlet right ventricle (DORV) with transposition of the great arteries accounts for 26% of cases of DORV. The aorta (AO) is anterior and to the right of the pulmonary artery (PA), and both arteries arise from the right ventricle (RV). The only outflow from the left ventricle (LV) is a ventricular septal defect (VSD), which diverts blood toward the RV. Pulmonary veins drain into the left atrium (LA) after blood has been oxygenated in the lungs (L). Systemic venous return is to the right atrium (RA).
-
This is an angiogram obtained during catheterization of a patient with double outlet right ventricle (DORV) with transposition of the great arteries. Injection of contrast though the catheter (arrow) into the left ventricle (LV) shows that blood is directed toward the right ventricle (RV) through a remote or doubly committed ventricular septal defect (VSD). The aorta (AO) is anterior to the pulmonary artery (PA) and both clearly arise from the RV.
-
This is an angiogram obtained during catheterization of a patient with double outlet right ventricle (DORV) with transposition of the great arteries (see Media file 2). Blood fills the aorta (AO) and pulmonary artery (PA) almost simultaneously, which is another indicator of a remote or doubly committed ventricular septal defect (VSD) (curved arrow). LV indicates the left ventricle, RV indicates the right ventricle, and the small arrow to the left indicates the catheter.
