Practice Essentials
Acute lymphoblastic leukemia (ALL) is the most common malignancy diagnosed in children, representing more than a quarter of all pediatric cancers. The image below depicts bone marrow aspirate from a child with T-cell acute lymphoblastic leukemia.
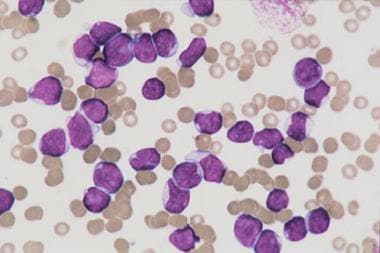 Bone marrow aspirate from a child with T-cell acute lymphoblastic leukemia. The marrow is replaced with lymphoblasts of various sizes. No myeloid or erythroid precursors are seen. Megakaryocytes are absent.
Bone marrow aspirate from a child with T-cell acute lymphoblastic leukemia. The marrow is replaced with lymphoblasts of various sizes. No myeloid or erythroid precursors are seen. Megakaryocytes are absent.
See Chronic Leukemias: 4 Cancers to Differentiate, a Critical Images slideshow, to help detect chronic leukemias and determine the specific type present.
Also, see the Childhood Acute Lymphoblastic Leukemia: Diagnosis, Management, and Complications slideshow to help recognize and treat this disease and its associated complications.
Signs and symptoms
Children with acute lymphoblastic leukemia (ALL) often present with signs and symptoms that reflect bone marrow infiltration and/or extramedullary disease. When leukemic blasts replace the bone marrow, patients present with signs of bone marrow failure, including anemia, thrombocytopenia, and neutropenia.
Other presenting signs and symptoms of pediatric ALL include the following:
-
Patients with B-precursor ALL: Bone pain, arthritis, limping; fevers (low or high); neutropenia; fatigue, pallor, petechiae, and bleeding; lymphadenopathy and hepatosplenomegaly
-
Patients with mature-B ALL: Extramedullary masses in the abdomen or head/neck; CNS involvement (eg, headache, vomiting, lethargy, nuchal rigidity)
-
Patients with T-lineage ALL: Respiratory distress/stridor due to a mediastinal mass
Symptoms of CNS involvement are rarely noted at initial diagnosis but are more common in T-lineage and mature B cell ALL. [1] Testicular involvement at diagnosis is also rare; if present, it appears as unilateral painless testicular enlargement.
See Clinical Presentation for more detail.
Diagnosis
Testing
Complete morphologic, immunologic, and genetic examination of the leukemic cells is necessary to establish the diagnosis of ALL.
Routine laboratory studies in pediatric ALL include the following:
-
Complete blood cell (CBC) count
-
Peripheral blood smear
-
Serum chemistries (eg, potassium, phosphorus, calcium)
-
Uric acid level
-
Lactate dehydrogenase (LDH) level
-
Coagulation studies, such as prothrombin time (PT), activated partial thromboplastin time (aPTT), levels of fibrinogen and D-dimer
Laboratory tests that help classify the type of ALL include the following:
-
Immunophenotyping - To detect surface immunoglobulin on leukemic blasts (diagnosis of mature B-cell leukemia) or the expression of T-cell–associated surface antigens (diagnosis of T-lineage ALL)
-
Cytogenetic studies - To identify specific genetic alterations in leukemic blasts
-
Molecular studies (eg, FISH, RT-PCR, Southern blot analysis) - To identify translocations more rapidly and those not detected on routine karyotype analysis; to distinguish lesions that appear cytogenetically identical but are molecularly different
-
Minimal residual disease studies [2] - To detect chimeric transcripts generated by fusion genes, detect clonal TCR or immunoglobulin heavy-chain (IgH) gene rearrangements, or identify a phenotype specific to the leukemic blasts on flow cytometry
-
Genome-wide association studies - To detect the presence of genetic changes where targeted therapy may be beneficial (eg, activated tyrosine kinase pathways in Ph-like ALL), not in clinical use yet
See Acute Lymphoblastic Leukemia Staging for more complete information.
Imaging studies
No other imaging studies other than chest radiography to evaluate for a mediastinal mass are routinely required in pediatric ALL. However, the following radiologic studies can be helpful:
-
Ultrasonography: To evaluate for testicular infiltration in boys with enlarged testes; to evaluate for leukemic kidney involvement as a risk assessment for tumor lysis syndrome
-
ECG, echocardiogram: To identify any preexisting cardiac dysfunction before administration of anthracyclines (baseline studies); to monitor heart function during treatment with anthracyclines
Procedures
-
Lumbar puncture with cytospin morphologic analysis: To assess for CNS involvement before administration of systemic chemotherapy; to administer intrathecal chemotherapy
-
Bone marrow aspiration and biopsy: To confirm the diagnosis of ALL
CNS disease is divided into the following groups:
-
CNS 1: Absence of blasts on CSF cytospin preparation, regardless of the WBC count
-
CNS 2: WBC count of less than 5/mL and blasts on cytospin findings, or WBC count of more than 5/mL but negative by Steinherz-Bleyer algorithm findings (if traumatic tap)
-
CNS 3: WBC count of 5/mL or more and blasts on cytospin findings and/or clinical signs of CNS leukemia (eg, facial nerve palsy, brain/eye involvement, hypothalamic syndrome)
See Workup for more detail.
Management
Leukemia is a systemic disease, and treatment is primarily based on chemotherapy. However, the different types of ALL require different approaches for optimal results. Treatment of subclinical CNS leukemia is an essential component of ALL therapy.
Treatment for ALL typically consists of the following phases:
-
Remission-induction phase (eg, dexamethasone or prednisone, vincristine, asparaginase, daunorubicin)
-
Intensification/consolidation phase: The importance of this phase is undisputed, but consensus is scarce on the best regimens and duration of treatment. Current Children's Oncology Group (COG) ALL protocols use a therapeutic backbone that was originally introduced in Berlin-Frankfurt-Muenster (BFM) clinical trials in the 1980s. This includes administration of cytarabine, cyclophosphamide, dexamethasone, asparaginase, doxorubicin, MTX, 6-MP, 6-thiouguanine, and vincristine.
-
CNS-directed therapy consists of systemic chemotherapy that enters the CSF, as well as intrathecal MTX administered throughout the entire course of treatment. Although this can include hydrocortisone and cytarabine (“triple-intrathecal therapy” or TIT), the COG High-Risk B-cell ALL trial (AALL1131) failed to show that TIT was more effective in preventing CNS relapse. [3]
-
Continuation therapy targeted at eliminating residual disease (eg, MTX, 6-MP, vincristine and glucocorticoid pulses)
Pharmacotherapy
Medications used in the treatment of pediatric ALL include the following:
-
Antineoplastics (eg, vincristine, asparaginase Escherichia coli, asparaginase Erwinia chrysanthemi, calaspargase pegol, daunorubicin, doxorubicin, MTX, 6-MP, cytarabine, cyclophosphamide, dasatinib, imatinib)
-
Corticosteroids (eg, prednisone, dexamethasone)
-
Antimicrobials (eg, TMX/SMP, pentamidine)
-
Antifungals (eg, fluconazole)
Clinical trial data from COG study AALL0434 showed that disease-free survival in T-cell ALL was superior with sequential escalating methotrexate ("Capizzi methotrexate"), and the addition of nelarabine, but different study groups still approach the management of T-cell ALL differently. Mature B-cell ALL is treated in the same way as disseminated Burkitt lymphoma, with short-term intensive chemotherapy, including high-dose MTX, cytarabine, and cyclophosphamide over a 6-month period.
Blood transfusions or antibiotics may be required to deal with complications of ALL therapy. Do not administer folate supplementation owing to interactions with MTX.
Nonpharmacologic therapy
Other treatments involved in managing pediatric ALL may include the following:
-
Initial administration of IV fluids: Without potassium, with or without sodium bicarbonate
-
Cranial irradiation: Effectively prevents overt CNS relapse but potentially causes neurotoxicity and brain tumors; largely replaced by intensive intrathecal and systemic chemotherapy
-
Allogeneic HSCT (hematopoietic stem cell transplant): Usually following second complete remission after relapse (if early) or first remission in high risk patients; potentially prevents relapse and/or mortality vs chemotherapy alone.
Surgical options
In generally, surgical care is not required in the treatment of ALL. However, placement of a central venous catheter is needed for administering chemotherapy, blood products, and antibiotics, as well as for obtaining blood samples.
See Treatment and Medication for more detail.
Background
Acute lymphoblastic leukemia (ALL) is the most common malignancy diagnosed in children, representing one quarter of all pediatric cancers. The annual incidence of acute lymphoblastic leukemia within the United States is 3.7-4.9 cases per 100,000 children age 0-14 years, [4] with a peak incidence in children aged 2-5 years.
Although a few cases are associated with inherited genetic syndromes (eg, Down syndrome) or congenital immunodeficiencies (eg, Wiskott-Aldrich syndrome, ataxia-telangiectasia), the cause remains largely unknown. [5]
With improvements in diagnosis and treatment, overall cure rates for children with acute lymphoblastic leukemia have reached 90%. [6] The use of risk-adapted treatment protocols has improved cure rates while limiting the toxicity of therapy. This article summarizes the current diagnosis and treatment of childhood acute lymphoblastic leukemia.
Pathophysiology
In acute lymphoblastic leukemia (ALL), a lymphoid progenitor cell becomes genetically altered and subsequently undergoes dysregulated proliferation, with clonal expansion. In ALL, the transformed lymphoid cells reflect the altered expression of genes usually involved in the normal development of B cells and T cells. Several studies indicate that leukemic stem cells are present in certain types of ALL.
Epidemiology
Annually, around 3000 children in the United States are diagnosed with ALL. The annual incidence of ALL within the United States is 3.7-4.9 cases per 100,000 children 0-14 years of age [4] with a similar estimated worldwide incidence, although it has been questioned whether the incidence may be less in low-income countries. [7] White children are more frequently affected than Black children, and there is a slight male preponderance, which is most pronounced for T-cell acute lymphoblastic leukemia. The incidence of acute lymphoblastic leukemia peaks in children aged 2-5 years and subsequently decreases with age.
Although a few cases are associated with inherited genetic syndromes (eg, Down syndrome) or congenital immunodeficiencies (eg, Wiskott-Aldrich syndrome, ataxia-telangiectasia), the cause remains largely unknown. [5] Environmental risk factors such as exposure to ionizing radiation and electromagnetic fields and parental use of alcohol and tobacco have not been shown to cause pediatric acute lymphoblastic leukemia. In addition, no direct link has been established between viral exposure and the development of childhood leukemia.
Prognosis
The likelihood of long-term cure in ALL depends on the clinical and laboratory features and the treatment. Prognostic risk assessment includes clinical features (age and white blood cell [WBC] count at diagnosis), biologic characteristics of the leukemic blasts, response to the induction chemotherapy, and minimal residual disease (MRD) burden. Based on these criteria, patients can be effectively stratified into low risk, average or standard risk, high risk, and very high-risk. [8]
Standard-risk patients are aged 1-9.9 years with a WBC count of less than 50,000 at presentation, lack unfavorable cytogenetic features, and show a good response to initial chemotherapy. The Children’s Oncology Group (COG) defines standard risk as less than 1% blasts in peripheral blood by 8 days and less than 0.01% blasts in bone marrow by 29 days (rapid early response). Low-risk patients have < 0.01% blasts for both time points and have favorable cytogenetics (eg, trisomy 4, 10). High-risk patients do not meet these criteria or have extramedullary involvement that makes it inappropriate for them to be treated as standard risk. Very-high-risk patients have unfavorable cytogenetic features (Philadelphia chromosome), hypodiploidy (n < 44), MLL gene rearrangement, or poor response to initial chemotherapy (induction failure or Day 29 bone marrow with MRD >0.01%).
Patients younger than 1 year with acute leukemia have disease that is biologically distinct with a poor outcome. [9]
The 5-year event-free survival (EFS) varies considerably depending on risk category, from 95% (low risk) to 30-80% (very high risk), with infant leukemia having the worst outcomes: 20% for patients younger than 90 days. COG redefined very high risk to include high-risk patients ≥13 years of age, which made the range of outcomes wider for this subgroup. Overall, the cure rate for childhood ALL is more than 80%.
Five-year survival rates for children diagnosed with ALL rose to 90% from 2000-2005, which was up from 84% in 1990-1994. [6] Improvement in survival was observed for all age groups of children, except for infants younger than 1 year. In low-income countries (LIC), therapeutic results for pediatric ALL have been less encouraging due to delayed diagnosis, abandonment of therapy, and death from toxicity due to suboptimal supportive care. Nevertheless, current 4-year event-free survival rates are 61% in India, [9] and over 78% in Lebanon, [10] demonstrating that pediatric ALL is curable in LIC.
An analysis of long-term survival among 21,626 children with ALL treated in COG trials from 1990-2005 found that 10-year survival rose to almost 84% in 1995-1999 from 80% in 1990-1994. The analysis also found that survival improved for almost all groups, including older children and Black children. [6]
Acute complications may involve all organ systems and include the following:
-
Tumor lysis syndrome
-
Renal failure
-
Sepsis
-
Bleeding
-
Thrombosis
-
Typhlitis
-
Neuropathy
-
Encephalopathy
-
Seizures
In addition, lifelong follow-up is necessary, because survivors may experience late effects from treatment for this condition, such as the following: [1]
-
Secondary malignancy
-
Short stature (if craniospinal radiation)
-
Growth hormone deficiency
-
Learning disability
-
Cognitive defects
Patient Education
Ensure that the patient's parents and guardians understand that ALL usually does not have a known cause, that accurate stratification helps guide therapy, and that participating in institutional or consortium-based clinical trials may help lead to better outcomes in the future. In addition, parents and guardians must know the expected adverse effects of each medication and be able to recognize signs and symptoms that require immediate medical attention, such as those for anemia, thrombocytopenia, and infection. Furthermore, parents and patients must know how to quickly access medical help from the oncology team.
For patient education information, see Cancer and Tumors Center, as well as Leukemia.
-
Bone marrow aspirate from a child with B-precursor acute lymphoblastic leukemia. The marrow is replaced primarily with small, immature lymphoblasts that show open chromatin, scant cytoplasm, and a high nuclear-cytoplasmic ratio.
-
Bone marrow aspirate from a child with T-cell acute lymphoblastic leukemia. The marrow is replaced with lymphoblasts of various sizes. No myeloid or erythroid precursors are seen. Megakaryocytes are absent.
-
Bone marrow aspirate from a child with B-cell acute lymphoblastic leukemia. The lymphoblasts are large and have basophilic cytoplasm with prominent vacuoles.
