Practice Essentials
Neural tube defects (NTDs) are significant birth deformities of the central nervous system that occur due to a defect in the neurulation process of embryogenesis. They are among the most common type of birth defects and are thought to have multifactorial etiology, including multigenetic and environmental influences. [1]
NTDs can be classified as “open” or “closed” types, based on embryological considerations and the presence or absence of exposed neural tissue (ie, failure of incomplete fusion of the neural plate).
-
Open NTDs frequently involve multiple aspects of the CNS (eg, associated hydrocephalus, Chiari II malformation) and are due to failure of primary neurulation, thus the neural tube fails to appropriately close along the dorsal midline. Neural tissue is completely exposed, or covered by a membrane, with associated cerebrospinal fluid (CSF) leakage. [2] Open NTD’s represent roughly 80% of all NTD’s, with the most common being meningocele (spina bifida), myelomeningocele, encephalocele, and anencephaly.
-
Closed NTDs are localized and confined to the spine (the brain is rarely affected) and result from a defect in secondary neurulation. Neural tissue is not exposed and the defect is fully covered by epithelium, although the skin covering the defect may be dysplastic (ie, tuft of hair, dimple, birthmark, or other superficial abnormality). [3]
Signs and symptoms
Most open NTDs are readily apparent at the time of birth; closed NTDs have a variable presentation.
The most common presentation of a closed NTD is an obvious abnormality along the spine such as a fluid-filled cystic mass, area of hypopigmentation or hyperpigmentation, cutis aplasia, congenital dermal sinus, capillary telangiectasia/hemangioma, hairy patch (hypertrichosis), skin appendages, or asymmetrical gluteal cleft. [4]
Diagnosis
Ultrasonography is used antenatally for NTD screening.
Maternal serum alpha-fetoprotein can be measured in maternal serum (MSAFP), amniotic fluid, and fetal plasma. It is typically measured around 16–18 weeks' gestation. MSAFP is a fetal-specific protein synthesized by the fetal yolk sac, GI tract, and liver. Interpretation of results varies by institution, but typically levels 2–2.5-fold above average (for a particular gestational age) is considered abnormal. However, many factors, both fetal and maternal, can affect interpretation of results. Abnormal MSAFP tests are typically followed by an ultrasound exam to assess for possible NTD, confirm gestational age, fetal viability, number of fetuses, and so on.
Management
Neurosurgical intervention is the mainstay of treatment for open neural tube defects (NTDs); closed NTDs typically do not warrant urgent surgery.
For open defects, prompt closure of the defect is indicated, ideally within the first 72 hours after birth for myelomeningocele. The closure involves classic neurosurgical techniques, involving the approximation of the lateral edges of the open neural plate to form a neural tube; this covers the open caudal end of the spinal cord with a layer of pia mater. [5] For closed defects associated with cord tethering, surgery involves removal of structures that are anchoring the cord.
Background
Neural tube defects (NTD) are significant birth deformities of the central nervous system that occur due to a defect in the neurulation process of embryogenesis. They are among the most common type of birth defects and are thought to have multifactorial etiology, including multigenetic and environmental influences. Because the neural tube is ultimately formed from the migration and fusion of the neural plate, the type and severity of malformation varies based on the location of the defect. This includes both cranial and spinal cord malformations. Since rostral and caudal neuropore closure is the last phase of neurulation, they are particularly vulnerable to defects. Consequently, a majority of NTDs arise in these areas. [6, 7]
NTDs can be classified as “open” or “closed” types, based on embryological considerations and the presence or absence of exposed neural tissue (ie, failure of incomplete fusion of the neural plate).
-
Open NTDs frequently involve multiple aspects of the CNS (eg, associated hydrocephalus, Chiari II malformation) and are due to failure of primary neurulation, thus the neural tube fails to appropriately close along the dorsal midline. Neural tissue is completely exposed, or covered by a membrane, with associated cerebrospinal fluid (CSF) leakage. [2] Open NTD’s represent roughly 80% of all NTD’s, with the most common being meningocele (spina bifida), myelomeningocele, encephalocele, and anencephaly.
-
Closed NTDs are localized and confined to the spine (the brain is rarely affected) and result from a defect in secondary neurulation. Neural tissue is not exposed and the defect is fully covered by epithelium, although the skin covering the defect may be dysplastic (ie, tuft of hair, dimple, birthmark, or other superficial abnormality). [3]
Cranial manifestations include the following: [8]
-
Anencephaly
-
Encephalocele (meningocele or meningomyelocele)
-
Craniorachischisis totalis
-
Congenital dermal sinus
Spinal presentations include the following: [8]
-
Spina bifida occulta
-
Spina bifida in relation to a dermoid cyst
-
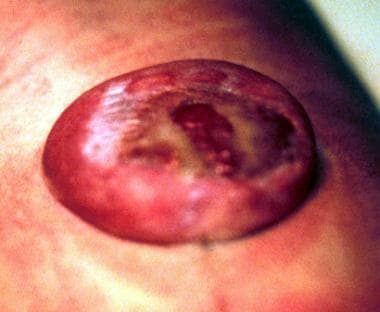
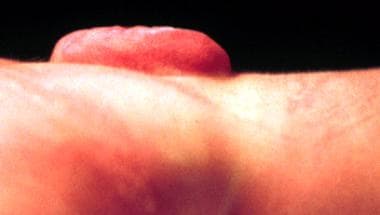
Spina bifida aperta (meningocele, myelomeningocele (see images below), meningomyelocele, myeloschisis)
-
Split-cord malformations
-
Diastematomyelia
-
Diplomyelia
-
Caudal agenesis
-
Lipomatous malformations (lipomyelomeningocele)
For more information on the classification of neural tube defects, see Medscape Reference article Imaging in Spinal Dysraphism and Myelomeningocele.
Pathophysiology
Two distinct and critical processes are involved in the formation of the neural tube: primary neurulation and secondary neurulation (i.e., canalization). [9] The neural plate and the notochord are formed during early embryonic development. The neural groove develops by the third gestational week and the neural folds subsequently form bilaterally.
Primary neurulation (weeks three and four during embryogenesis, forming the early brain and spinal cord): [10]
-
The neural folds elevate, approximate each other, and start fusing along the dorsal midline, thus forming the neural tube. This begins during the third week after conception.
-
The cranial neuropore closes during the fourth gestational week. The last area to close is the commissural plate.
-
The point of initial closure occurs at the caudal rhombencephalon, or cranial neuropore, on day 25 of embryogenesis.
-
The cutaneous ectoderm fuses first, followed by the neuroectoderm.
-
The caudal neuropore closes between T11 and S2, two days after closure of the cranial neuropore (roughly day 27)
-
Occurring in parallel to this process, the cutaneous ectoderm separates from the neuroectoderm to form the overlying skin, while the lateral mesoderm migrates between the 2 ectodermal layers to form the posterior vertebral arches.
Secondary neurulation (canalization: weeks five and six, forming the early sacral and coccygeal cord)
-
This characterizes further neural development caudal to the caudal neuropore after the termination of primary neurulation, and the caudal neuropore closes
-
This process includes formation of the filum terminale and conus medullaris from a poorly differentiated cell mass of the medial eminence.
-
Because of differential growth between the vertebral column and the spinal cord, the conus becomes more rostral during later development.
-
By 8 weeks after conception, spinal cord tissue runs the entire length of the spinal cord. [11]
Open NTDs have been suggested to result from defective primary neurulation while defective secondary neurulation gives rise to closed NTDs. However, this issue is not settled. Another possible explanation is that open NTDs (spina bifida in particular) result from defects in either primary or secondary neurulation, depending on their site being cranial or caudal to the posterior neuropore (ie, upper and lower spina bifida, respectively).
Epidemiology
Epidemiological studies have demonstrated multifactorial influences underlying neural tube defects (NTDs). Variations in incidence have been seen across geographical variation, ethnic groups, socioeconomic status, and genetic predisposition.
Frequency
In the United States, spina bifida affects about 1300 babies a year and anencephaly affects about 700 babies each year. Encephalocele is a rarer NTD, affecting about 350 babies each year nationwide. [12]
NTDs are among the most common birth defects globally, affecting more than 300,000 births annually. [13, 1] Estimates vary widely by country and WHO region, and there is a lack of high-quality data from regions with the highest burden. The prevalence of NTDs in the Western Pacific region ranges from 0.3 to 199.4 per 10,000 births, while in Europe it ranges from 1.3 to 35.9 per 10,000 births.
A 2016 study found the following ranges and medians per 10,000 births: [14]
-
African: 5.2–75.4, median of 11.7
-
Eastern Mediterranean: 2.1–124.1, median of 21.9
-
European: 1.3–35.9, median of 9.0
-
Americas: 3.3–27.9, median of 11.5
-
South-East Asian: 1.9–66.2, median of 15.8
-
Western Pacific: 0.3–199.4, median of 6.9
Mortality/morbidity
In the United States, the infant mortality rate due to NTDs was 8.3 per 100,000 live births between 2018 and 2021. However, with new treatments, surgeries, and therapies, at least 75% of people with spina bifida can live into adulthood. [12]
Anencephaly is incompatible with life. [15] No differentiated supratentorial neural tissue is present, and the brain stem consists of nests of poorly differentiated neural elements.
The brain stem is believed by some to be not sufficiently developed to be responsible for the temporary brainstem reflexes that are observed. Some have implicated the upper cervical cord as the seat of these functions.
The survival of these newborns is limited to a few hours (rarely >2 days).
Other NTDs may give rise to progressive neurological deterioration, which may present early after birth or later in life. The neurological deficits may be due to accompanying hydrocephalus, a Chiari II malformation, tethering of the cord, cystic mass, or fibrous band compressing the neural elements. Another possible complication is meningitis (infectious or chemical), especially in open NTDs.
The average recurrence risk of NTDs for parents with one affected child has been estimated to be about 5%, and that for monozygotic twins about 20%. Recurrence risks are higher in populations with a higher birth incidence.
The most common NTD compatible with life and a positive prognosis is myelomeningocele (see the images below).
The incidence of myelomeningocele is about 3.5 cases per 1000 live births (1400–1645 infants annually). [16]
Paralysis, bladder and bowel incontinence, and hydrocephalus are the most common clinical complications. Severe intellectual disability is present in 10–15% of these patients.
Neurologic deficits are overall difficult to predict based on the level of the lesion, as some segments of the spinal cord may retain central connections and maintain partial function, allowing voluntary control or sensation in affected limbs. Most commonly, distal cord may retain function, but afferent pathways are interrupted. This may preserve reflexes and pain withdrawal, but voluntary movement and pain sensation are affected.
Prospects for independent ambulation are correlated with the level of the spinal lesion. Independent mobility is preserved for nearly all cases with low lumbar and sacral lesions; with lesions above L2, loss of quadriceps and iliopsoas muscle function often occurs, and independent mobility is unlikely. [17]
Bowel and bladder function are affected in roughly 90–95% of patients with myelomeningocele, manifesting as neurogenic bladder and/or fecal incontinence. [18]
Presence or severity of urinary dysfunction cannot be predicted by location of the spinal lesion or by neurologic exam. The level of spinal cord responsible for mediating bladder function is below the level that controls lower extremity function. [19]
Despite aggressive medical care, 10–15% of these children die prior to reaching the first grade. However, most children with isolated myelomeningocele (without major anomalies of other organs) survive to adulthood, and life expectancy is nearly normal. [20] Sixty percent have normal intelligence, although of these, 60% have some learning disability (math and problem solving being particularly difficult). Attention deficit disorder without hyperactivity also has been described in these children. Hydrocephalus is present in 85% but bears little relationship to intelligence. About 80% are socially continent (although many require clean intermittent catheterization).
Race
In studies done before the availability of prenatal screening and prophylactic vitamin supplementation, birth incidence of both spina bifida and anencephaly was reported as higher in the European white population than in the black population. [13]
In North America, the risk of NTDs was found to be highest in the Hispanic population (more than 3-fold higher than that for non-Hispanic whites).
Migration studies in the white migrant population showed a prevalence of NTDs that corresponded more closely to the risk of the place to which they had migrated, as opposed to the place of their origin. In contrast, similar studies in descendants of the Black and Asian migrant populations in Europe and North America showed a prevalence not substantially higher than those of their parent countries. These variations are consistent with the theory that NTDs are a phenotypically heterogeneous group of malformations with environmental factors, multifactorial inheritance in some cases, and single gene defects in others.
Sex
Anencephaly has a female preponderance, especially among premature births, with a female-to-male ratio of 3:1.
Other NTDs above the thoracolumbar junction show a mild female preponderance.
No such gender difference has been noted in more distal forms of spina bifida.
Age
Open NTDs are readily visible at birth, with the majority being discovered during pregnancy.
Closed NTDs may remain undetected for years, even decades, especially in the absence of cutaneous markers; it has been estimated that roughly 70% of asymptomatic patients with a closed NTD present with one or more cutaneous lesion. [21, 22]
Prognosis
Prognosis in patients with neural tube defects (NTDs) depends upon the defect and ranges from excellent to poor.
-
Myelomeningocele in a newborn.
-
Myelomeningocele in a newborn - Lateral view.
-
Child with Chiari malformation, in whom the tonsils have descended to the level of C2.
-
MRI of a cervical syrinx in the sagittal plane.
-
MRI of a cervical syrinx in the axial plane.
