Background
X-linked agammaglobulinemia (XLA), or Bruton agammaglobulinemia, is an inherited immunodeficiency disease caused by mutations in the gene coding for Bruton tyrosine kinase (BTK). The disease was first elucidated by Bruton in 1952, for whom the gene is named. BTK is critical to the maturation of pre–B cells to differentiating mature B cells. The BTK gene defect has been mapped to the long arm of the X chromosome at band Xq21.3 to Xq22, spanning 37.5kb with 19 exons forming 659 amino acids to complete the BTK cytosolic tyrosine kinase. A TH domain missense mutation has also been described in BTK. [1] A database of BTK mutations (BTKbase: Mutation registry for X-linked agammaglobulinemia) lists 544 mutation entries from 471 unrelated families showing 341 unique molecular events. No single mutation accounts for more than 3% of mutations in patients. In addition to mutations, a number of variants or polymorphisms have been found. See the image below.
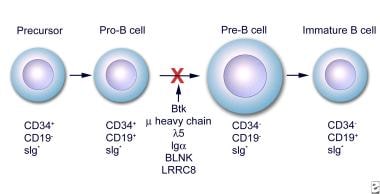 Early stages of B-cell differentiation can be identified by the status of the immunoglobulin genes and by the cell surface markers CD34, CD19, and surface immunoglobulin (sIg). From: Conley ME. Genes required for B cell development. J Clin Invest. 2003;112: 1636-8. Reproduced with permission of American Society for Clinical Investigation via Copyright Clearance Center.
Early stages of B-cell differentiation can be identified by the status of the immunoglobulin genes and by the cell surface markers CD34, CD19, and surface immunoglobulin (sIg). From: Conley ME. Genes required for B cell development. J Clin Invest. 2003;112: 1636-8. Reproduced with permission of American Society for Clinical Investigation via Copyright Clearance Center.
The Medscape Reference Pediatrics article, Bruton Agammaglobulinemia, also may be of interest.
Pathophysiology
In the absence of BTK, B lymphocytes do not differentiate or mature. Without mature B lymphocytes, antibody-producing plasma cells are also absent. As a consequence, the reticuloendothelial and lymphoid organs in which these cells proliferate, differentiate, and are stored are poorly developed. The spleen, the tonsils, the adenoids, the Peyer patches in the intestines, and the peripheral lymph nodes may all be reduced in size or absent in individuals with X-linked agammaglobulinemia (XLA).
The protooncogene encoding for BTK has been cloned and its genomic organization determined, allowing an in-depth analysis of the role of BTK and other signaling molecules in B-cell differentiation. [2, 3]
Mutations in each of the 5 domains of BTK can lead to disease. The single most common genetic event is a missense mutation. Most mutations lead to truncation of the BTK enzyme. These mutations affect critical residues in the cytoplasmic BTK protein and are highly variable and uniformly dispersed throughout the molecule. Nevertheless, the severity of disease cannot be predicted by the specific mutations. Approximately one third of point mutations affect CGG sites, which usually code for arginine residues. The putative structural implications of all of the missense mutations are provided in the database. [4, 5, 6, 7]
BTK is necessary for the proliferation and the differentiation of B lymphocytes. [8, 9, 10] Males with XLA have a total or almost total absence of B lymphocytes and plasma cells. XLA is an inherited disease that occurs in approximately 1 in 250,000 males. Female carriers have no clinical manifestations. Infections begin once transferred maternal immunoglobulin G (IgG) antibodies have been catabolized, typically at about 6 months of age.
Diagnosis
Early detection and diagnosis is essential to prevent early morbidity and mortality from systemic and pulmonary infections. The diagnosis is confirmed by abnormally low or absent numbers of mature B lymphocytes, as well as low or absent expression of the µ heavy chain on the surface of the lymphocyte. Conversely, T-lymphocyte levels are elevated. The definitive determinant of XLA is the complete absence of BTK ribonucleic acid (RNA) or protein. Specific molecular analysis is made by single-strand confirmation polymorphism (SSCP), direct DNA analysis, denaturing gradient gel electrophoresis, or reverse transcriptase–polymerase chain reaction to search for the BTK mutation. SSCP is also used for prenatal evaluation, which can be performed via chorionic villus sampling or amniocentesis when a mother is known to be a carrier. IgG levels less than 100 mg/dL support the diagnosis.
Rarely, the diagnosis is made in adults in their second decade of life. This is thought to be due to a mutation in the protein, rather than a complete absence.
Medicolegal concerns may include the following:
-
Failure to diagnose XLA in a male with a documented family history of the disease
-
Failure to interpret relative laboratory tests, such as immunoglobulin levels or antibody responses
-
Failure of the family and medical personnel to monitor intravenous immunoglobulin (IVIG) infusions
-
Failure of the physician to withhold all live viral vaccines
-
Failure of a physician to educate a patient with XLA about health care and maintenance
Etiology
The BTK mutations underlying X-linked agammaglobulinemia (XLA) interferes with the development and the function of B lymphocytes and their progeny. The major block occurs in the development of pro–B cells to pre–B cells and then to mature lymphocytes. Patients can have pre–B cells in the marrow, but they have few, if any, functional (mature) B cells in the peripheral blood and the lymphoid tissues.
Epidemiology
Frequency
United States
The estimated frequency of X-linked agammaglobulinemia (XLA) is approximately 1 case per 250,000 population. Two thirds of cases are familial, and one third of cases are believed to arise from new mutations.
International
The incidence of XLA around the world does not vary significantly.
Race
Most studies involve Northern European patients. However, no racial predilection for XLA has been established.
Sex
Bruton agammaglobulinemia is an X-linked disease, with only male offspring being affected. Most cases are inherited, but, rarely, the disease manifests as a consequence of a spontaneous mutation. Mutations in the gene for the heavy mu gene (IGHM), the immunoglobulin-alpha gene, and the lambda-5 gene can cause agammaglobulinemia, with less than 1% CD19 expression on B cells. No female carriers present with the clinical manifestations of the BTK mutation.
Age
Male infants become affected by X-linked agammaglobulinemia (XLA) when maternal antibodies decline usually after age 4-6 months. If the mother has been identified as a carrier for the disease, chorionic villi sampling or amniocentesis can be performed to collect fetal lymphocytes in utero. At birth, cord blood samples can be tested for a decrease in CD19+ B cells and for an increase in mature T cells via fluorocytometric analysis. Children typically clinically manifest the disease at age 3-9 months with pneumonia, otitis media, cellulitis, meningitis, osteomyelitis, diarrhea, or sepsis. [11, 12] Rare cases of adults in their second decade have been diagnosed with a milder form XLA thought to be due to a mutation rather than an absence of the protein.
Prognosis
Patients with XLA have survived into their late 40s. The prognosis is good as long as patients are diagnosed and treated early with regular intravenous gamma globulin therapy before the sequelae of recurrent infections appear. [13]
IVIG is responsible for increasing survival rates, with treatment beginning preferably before the patient is aged 5 years.
Serious enteroviral infections and chronic pulmonary disease are often fatal in adulthood.
Mortality/morbidity
Most men with X-linked agammaglobulinemia (XLA) live into their 40s. The prognosis is better if treatment is started early, ideally if intravenous immunoglobulin G (IVIG) is started before the individual is aged 5 years. Even with treatment, patients can expect to have chronic pulmonary infections, skin disease, inflammatory bowel disease (ulcerative colitis and Crohn disease), and central nervous system complications due to enteroviral infection.
Patient Education
Patients and their families must understand the nature of the disease and the importance of early treatment. Identification and treatment of common infections are necessary for a better prognosis.
Genetic counseling is recommended for the parents and female siblings of males who are affected. Molecular characterization and carrier detection is informative in 95% of families. Prenatal diagnosis is available. [14, 15] A combination of flow cytometry and Bruton tyrosine kinase gene analysis may be beneficial for carrier screening. [16]
The Immune Deficiency Foundation is a solid resource for both support and education of patients and their families. The foundation can be reached at 1-800-296-4433. The Jeffery Modell Foundation can be reached at 1-800-JEFF-844.
-
Early stages of B-cell differentiation can be identified by the status of the immunoglobulin genes and by the cell surface markers CD34, CD19, and surface immunoglobulin (sIg). From: Conley ME. Genes required for B cell development. J Clin Invest. 2003;112: 1636-8. Reproduced with permission of American Society for Clinical Investigation via Copyright Clearance Center.
