Practice Essentials
Alpha thalassemia is a widespread, inherited anemia that results from impaired production of α globin, a molecule essential to the synthesis of hemoglobin A (HbA, or α2β2) in red blood cells.
Signs and symptoms
Symptoms vary markedly depending on the number of impaired α-globin genes; since healthy adults have four functional α-globin genes (two on each allele), losing one or two of them usually has minimal or no impact, as seen in the following [1] :
-
Silent carriers - With a single α-globin gene impaired (-α/αα), these individuals are essentially asymptomatic
-
Alpha thalassemia trait - With two α-globin genes impaired (-α/-α or --/αα), individuals with this trait may have mild microcytosis and hypochromia, with borderline anemia
By contrast, losing three or four α-globin genes has a significant clinical impact, as seen in the following:
-
Hemoglobin H (HbH) disease (alpha thalassemia intermedia) - With three α-globin genes impaired, an excess of β-globin chains leads to HbH (β4) disease (--/-α), with affected individuals having anemia and splenomegaly. When the loss of these α-globin genes is by deletion (deletional HbH), the disease tends to be milder, whereas patients with inactivating point mutations of the α-globin gene (non-deletional HbH) commonly need red cell transfusions and may rarely manifest with HbH hydrops fetalis [2, 3]
-
Hemoglobin Bart hydrops fetalis (alpha thalassemia major) - With all four α-globin genes impaired (--/--), an inability to make functional hemoglobin leads to death in utero or shortly after birth for affected individuals, due to severe anemia with circulating Hb Bart (ϒ4); this anemia leads to placental enlargement, massive edema, congestive heart failure, and, as a result of heart failure and extramedullary hematopoiesis, massive hepatomegaly [2]
Workup and management
Investigations and management vary depending on the severity of the illness. For silent carriers and in alpha thalassemia trait, the focus is on obtaining an accurate diagnosis so that genetic counseling can be provided where appropriate. A simple blood count, red cell morphology, and reticulocyte count can usually clarify the probable diagnosis, with all tests in silent carriers and persons with alpha thalassemia trait being essentially normal except for very mild microcytic hypochromic anemia in patients with alpha thalassemia trait.
Hemoglobin electrophoresis and a serum ferritin level can help to exclude beta thalassemia trait and iron deficiency. Genetic testing is now available to establish the diagnosis and clarify the mutations present in patients with a family history or laboratory results suggestive of an alpha thalassemia syndrome, and polymerase chain reaction (PCR) assays or restriction endonuclease testing may be used. [4, 5, 6] Identifying carriers who have a high risk of having severely affected children has taken on a new urgency as populations migrate to various parts of the globe, such as the US state of California. [4]
Individuals with HbH disease (-α/--) have moderate to severe microcytic hypochromic anemia with Hb 7-10 g/dL, reticulocytosis of 5-10%, and HbH inclusion bodies, the latter being visible in RBCs on brilliant cresyl blue stain. Patients with more severe anemia in HbH disease may require intermittent or ongoing transfusion therapy, and these are usually patients with non-deletional HbH. Supplementation with folic acid may be useful. [3] While surgical care is not needed for silent carriers or persons with alpha thalassemia trait, splenectomy may be beneficial for more severely affected patients with HbH disease. In very severe cases of HBH disease, allogeneic hematopoietic stem cell transplantation (HSCT) may occasionally be an option. [7]
Individuals with Hb Bart hydrops fetalis (--/--) present a special challenge and are detected in utero with severe anemia, congestive heart failure, and hepatosplenomegaly, along with a massive placenta. While alpha thalassemia major is commonly fatal, in utero transfusion (IUT) and HSCT have allowed patients to survive. [8] Congenital anomalies may also occur, and the diagnosis may not be initially suspected in regions where alpha thalassemia carriers are rare, especially if the fetus is an early stillbirth. [4]
Alpha thalassemia trait can coexist with other hemoglobinopathies, with variable outcomes; for example, when occurring with sickle-cell disease, alpha thalassemia trait results in a higher hemoglobin concentration and improved RBC survival, but it may not affect overall patient survival.
Background
Thalassemia is one of the world’s most common single-gene disorders. The oxygen-carrying capability in RBCs relies on the protein hemoglobin. Hemoglobin A, the dominant hemoglobin in adults, is composed of a heme molecule bound to two globin chains produced by the α-globin gene locus and two globin chains from the β-globin gene locus (α2β2).
β-globin mutations are commonly found in populations residing around the Mediterranean, and in 1932 George Whipple from Rochester, NY, coined the term "thalassemia" for the inherited anemia observed in such individuals ("thalassa" = "great sea" in Greek). Severely impaired production of β-globin chains results in the disease known as beta thalassemia. [9] It is now known that beta thalassemia is found worldwide, including Iran, India, Southeast Asia, and China. While point mutations are the most common abnormality causing beta thalassemia, small deletions may also occur. Mutations and gene deletions causing thalassemia have arisen independently in different populations but have subsequently propagated by means of natural selection, possibly due to a protective effect against malaria. [10]
Alpha thalassemia is also found primarily in the Mediterranean, as well as in Africa and southern Asia and is characterized by impairment of one or more of the four α-globin genes needed to produce α-globin chains for fetal hemoglobin in utero (α2γ2) and adult hemoglobin (α2β2) following birth. If only one or two α-globin genes are affected, the impact is minimal, being silent or resulting in a very mild microcytic hypochromic anemia. More severe decrease in the production of α-globin chains yields a relative excess of β chains, resulting in the development of the less stable hemoglobin H (β4) and the clinical disease known as alpha thalassemia intermedia, or HbH disease.
Complete absence of α-globin chain production leads to the absence of normal circulating hemoglobins, with only Hb Bart (γ4), named after its place of discovery, St. Bartholomew's Hospital in London, produced in utero. This molecule is unable to effectively release oxygen in tissues, with the resulting disease, Hb Bart hydrops fetalis, usually resulting in stillbirth. [2, 4]
Pathophysiology
Genes that regulate the synthesis of different globins are organized into two separate clusters. The α-globin genes are encoded on chromosome 16, and the γ-, δ-, and β-globin genes are encoded on chromosome 11. Healthy individuals have four α-globin genes, two on each allele of chromosome 16 (αα/αα). The two α-globin genes on the same allele are referred to as α1 globin (HBA1) and α2 globin (HBA2), with the latter being responsible for 75% of α-globin chain production from that allele. [11] Alpha thalassemia syndromes result from deficient expression of one or more of the four α-globin genes on chromosome 16, causing reduced or absent synthesis of α-globin chains.
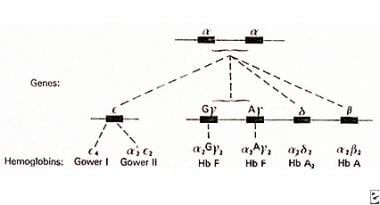 Alpha-chain genes in duplication on chromosome 16 pairing with non-α chains to produce various normal hemoglobins.
Alpha-chain genes in duplication on chromosome 16 pairing with non-α chains to produce various normal hemoglobins.
 The severity of alpha thalassemia is primarily dependent on the number of α-globin genes that have been deleted or inactivated, but it can be affected by co-existing factors as well.
The severity of alpha thalassemia is primarily dependent on the number of α-globin genes that have been deleted or inactivated, but it can be affected by co-existing factors as well.
While the terminology is occasionally confusing, in αo thalassemia (formerly called alpha thalassemia-1), production of α globin is absent from the allele due to large deletions that remove both of the α-globin genes present on that allele. There are over 40 different genetic mutations that result in the deletion of both α-globin genes on the same allele; they are characterized by their length or place/population of origin, with the most common being as follows [12] :
-
--MED Mediterranean
-
--SEA Southeast Asia
-
--FIL Filipino
-
--THAI Thailand
In the more widespread α+ thalassemia (formerly called alpha thalassemia-2), there is decreased production of α globin from a given allele, usually resulting from relatively small deletions of 3.7 kb or 4.2 kb that remove a single α-globin gene out of the two present on the allele. [1] It should be noted that in less than 10-25% of cases, α+ thalassemia results from a non-deletional α-globin mutation (written as αND or αT) and that this is sometimes associated with production of a hemoglobin variant. The best known example is Hb Constant Spring (HbCS), originally described in a Chinese family from Constant Spring, Jamaica, and widely prevalent in Southeast Asia, where the αCS mutation results in an elongated α chain and a down-regulated α2-globin gene. [12] Hemoglobins Adana and Quong Ze, which impair α-globin production, have also been described in Asia, [13] and other variants have been described in Saudi Arabia, Iran, and India. [14, 13, 15]

Silent carriers
While alpha thalassemia syndromes are genetically heterogeneous, the potential impact can be simplified to how many α-globin genes have been impaired. Persons who inherit three normal α-globin genes (-α/αα) are referred to as silent carriers. Other names for this condition are alpha thalassemia-2 trait and heterozygous α+-thalassemia minor. The affected individuals exhibit no clinical abnormalities and are hematologically normal.
Alpha thalassemia trait
Inheritance of two normal α-globin genes—through either heterozygosity, in αo thalassemia (--/αα), or homozygosity, in α+ thalassemia (-α/-α)—results in alpha thalassemia trait.
When both α-globin genes are deleted on the same chromosome (--/αα), the genotype is sometimes described as having the cis form; when one α-globin gene is deleted from each allele (-α/-α), the genotype is said to have the trans form. The affected individuals are clinically normal but frequently have minimal anemia and reductions in mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH). The RBC count is usually increased, typically exceeding 5.5 × 1012/L. The genetic implications for individuals who are αo-thalassemia heterozygotes is considerable, as offspring are at risk for severe disease.
HbH disease
Inheritance of three affected α-globin genes (-α/--) leads to HbH disease, in which an excess of β-globin chains forms soluble tetramers (β4), or HbH. HBH will in these cases account for 15-30% of circulating hemoglobin; HbH is unstable and precipitates within the red cells, forming Heinz bodies, which damage the red cell membrane. [16] HbH has a high affinity for oxygen, with no Bohr effect or heme-heme interaction, and is an ineffective supplier of oxygen, so that symptoms are more severe than might be expected from the patient's hemoglobin level.
Aging erythrocytes with precipitated HbH are removed from the circulation prematurely, making HbH disease primarily a hemolytic disorder. Unlike beta thalassemia, ineffective erythropoiesis is not a primary feature, but erythroid hyperplasia can still result in bone abnormalities with marrow hyperplasia, bone thinning, maxillary hyperplasia, and pathologic fractures.
It is well recognized that non-deletional HbH disease, in which a point mutation impacts at least one α-globin gene out of the three, is more clinically severe than the deletional form. Non-deletional HbH disease is commonly seen in Southeast Asia, where αo-thalassemia is co-inherited with HbCS, leading to the phenotypically severe HbH Constant Spring (HCS) disease. However, co-inheritance of other α-globin point mutations can also cause increased severity and in rare cases can even lead to HbH hydrops fetalis. [13, 3] Newborn screening can detect patients with HbH disease due to a relative excess of γ-globin chains, which leads to 15-30% circulating Hb Bart (γ4) at time of birth.
The results of one study suggested that HCS should be identified as a distinct thalassemia syndrome as it tends to be more severe, with a high risk of life-threatening anemia. [17]
Hb Bart hydrops fetalis
When αo thalassemia occurs in a homozygous state (--/--), then the resulting disorder is referred to as Hb Bart hydrops fetalis, or alpha thalassemia major. Individuals with this disorder cannot produce functional α-globin and are thus unable to make any functional hemoglobin A, F, or A2. Fetuses with this condition usually die in utero in the second or third trimester or shortly after birth, because of severe anemia. While the mortality rate for hydrops fetalis is high, medical advances have permitted some of these infants to survive, but aggressive intervention at specialized centers is needed.
Etiology
Normal hemoglobin biosynthesis requires an intact gene, silencers, enhancers, promoters, and locus control region (LCR) sequences. Several hundred mutations causing thalassemia have been described. These may affect any step in globin gene expression, transcription, pre-mRNA splicing, mRNA translation and stability, and posttranslational assembly and stability of globin polypeptides. Whereas beta thalassemia results from point mutations in the β-globin genes or from small deletions, the most common mechanism for aberrant α-globin production involves deletion of either portions of the α-globin genes themselves or the genetic regulatory elements that control their expression. Regulatory elements may be located on the same chromosome (cis-acting elements) or on separate chromosomes (trans-acting elements).
The (--SEA) type of alpha thalassemia deletion removes both α-globin genes on the same allele (in cis); it is common in Southeast Asia (hence its name) and is the most frequent cause of HbH disease and hydrops fetalis in that part of the world. [17] More extensive deletions that remove embryonic globin genes, such as (--FIL), common in the Philippines, are more likely to lead to stillbirth in the first trimester when inherited in the homozygous state. [18]
Non-deletional forms of alpha thalassemia, in which the α-globin genes are intact, are caused by point mutations; they are uncommon, accounting for less than 10-25% of alpha thalassemia carriers. [19, 15] In non-deletional mutations, production of functional hemoglobin is impaired when point mutations, frame shift mutations, nonsense mutations, and chain termination mutations occur within or around the coding sequences of the α-globin gene cluster. These gene-level mutations may in turn affect RNA splicing, hinder initiation of mRNA translation, or result in the generation of unstable α-globin chain variants.
Mutations affecting transcription, pre-mRNA splicing, or canonical splice signals are rare causes of alpha thalassemia. Other forms of alpha thalassemia are caused by either premature or failed translation termination. Even rarer mutations have been found to cause thalassemia by interfering with the normal folding of an otherwise normal globin peptide. [20]
Epidemiology
Alpha thalassemia is among the most common single-gene disorders in the world. It is estimated that there are 270 million carriers of mutant globin genes that can potentially cause severe forms of thalassemia. In addition, 300,000-400,000 severely affected infants are born every year, more than 95% of whom are in Asia, India, and the Middle East. [4]
Race/ethnicity
The frequency of alpha thalassemia is low among Whites, but it is estimated that about 15% of American Blacks are silent carriers of alpha thalassemia and that about 3% have alpha thalassemia trait; HbH disease is rare in the American Black population.
Alpha thalassemia occurs in individuals of all ethnic backgrounds but particularly in those of African, Asian, Central American, Mediterranean, and Middle Eastern descent. [21, 22, 23, 24, 25, 26] Emigration from regions in which carrier frequency is high increases the presence of thalassemia syndromes in other parts of the world. The National Institutes of Health (NIH)–sponsored North American Thalassemia Clinical Research Network (TCRN) study of the epidemiology of thalassemia in North America showed that 85% of patients with alpha thalassemia are Asian, 4% are white, and 11% are of other ethnicities, including African, black, mixed ethnicity, and unknown. [27, 28]
In some ethnic groups, such as the Chinese, the Southeast Asians (in particular), and the Mediterranean populations, HbH disease and Hb Bart hydrops fetalis are common because of the widespread prevalence of αo-thalassemia carriers. Frequent co-inheritance of an allele lacking both α-globin genes and another allele lacking one α-globin gene leads to HbH disease. The high frequency of hemoglobin Constant Spring (HbCS) in the Southeast Asian population can lead to the HbH (--/-αCS) phenotype, which involves an elongated form of alpha-globin chain. [3, 2]
According to the North American TCRN study, 59% of patients with alpha thalassemia have a single α-globin gene (-α/--), 8% have no α-globin genes (--/--), and 33% have gene deletions in combination with structural mutations. [27, 28] Before the introduction of DNA analysis, population surveys for alpha thalassemia were based entirely on the measurement of Hb Bart levels in cord blood. However, single-gene–deletion heterozygotes do not always have detectable Hb Bart in the neonatal period. As a result, reliable data on population frequencies for various types of alpha thalassemia are not always available. Alpha thalassemia is common throughout parts of the world where malaria is endemic. Multiple studies have suggested that the presence of both single and double α-globin gene deletions has a protective effect against malaria. [10, 29]
 This demonstrates α0 thalassemia resulting from deletions that remove large portions of DNA on the same allele (“in cis”), including, in some cases, the embryonic zeta-globin gene (eg, --THAI, --FIL).
This demonstrates α0 thalassemia resulting from deletions that remove large portions of DNA on the same allele (“in cis”), including, in some cases, the embryonic zeta-globin gene (eg, --THAI, --FIL).
The frequency of alpha thalassemia alleles is 5-10% in the Mediterranean basin, 20-30% in portions of West Africa, and as high as 60-80% in some populations in Saudi Arabia, India, Thailand, Papua New Guinea, and Melanesia. In Thailand, which has a population of 62 million people, approximately 7000 infants are born each year with HbH disease. The frequency of heterozygote carrier status among the Chinese population has been reported to range from 5-15%. The frequency of alpha thalassemia is lower than 0.01% in Great Britain, Iceland, and Japan. [30, 18, 12, 31]
Age
Abnormalities of α-globin chains are genetic, and individuals are born with the disorder. [32] The exception to this rule is patients with alpha thalassemia myelodysplastic syndrome (ATMDS), a form of HbH observed in myeloproliferative diseases (eg, acute myelogenous leukemia, erythroleukemia, refractory sideroblastic anemia, acute lymphocytic leukemia). Patients with ATMDS are usually elderly, with a mean age of 68 years at diagnosis. [33, 34]
Sex
In general, males and females are equally affected by alpha thalassemia. However, there is a type of alpha thalassemia that is associated with intellectual disability and affects only males. This condition is referred to as alpha thalassemia X-linked intellectual disability syndrome (ATR-X). However, a report by Haas et al identified 2 females in a single center with ATMDS and mutations in the ATR-X gene (ATRX). [33] The investigators observed that although it appeared possible that females might be less likely to develop ATMDS if reactivation of the inactivated copy of ATRX occurred throughout life, this hypothesis was ruled out in their study by the use of a cross-sectional analysis to examine the pattern of ATRX inactivation, looking at healthy females ranging in age from neonate to 90 years.
Prognosis
For silent carriers and individuals with alpha thalassemia trait, the prognosis is excellent.
For individuals with HbH disease, the overall survival rate varies but is generally good, but while most patients survive into adulthood, some patients have a more complicated course and may not do as well. Patients with HbH disease are at risk for severe anemia and have a lifelong requirement for transfusions. HbH disease with non-deletional mutations such as HbCS (HCS) is associated with a more severe phenotype and a high risk of life-threatening anemia. [17] Patients identified with non-deletional subtypes of HbH disease require especially close follow-up. If anemia is well managed and iron overload is prevented with chelation therapy, individuals with HbH disease can live long and healthy lives.
Hb Bart hydrops fetalis (alpha thalassemia major) is incompatible with life and requires identification in utero, with in utero transfusions if the fetus is to survive. To identify fetuses with this condition, family genetic studies must be done, high-risk couples identified, and the fetus tested in utero for the absence of α-globin chains. Those rare fetuses that survive with the help of intrauterine transfusions (IUTs) will continue to require lifelong transfusions and medical care and may be considered for hematopoietic stem cell transplantation (HSCT), which is curative for their disease.
A study by Joly et al supported the idea that alpha thalassemia reduces the risk for cerebral vasculopathy in children with sickle-cell anemia. [35]
A study by Brewin et al indicated that in persons with sickle-cell disease, either the HbSS or HbSC genotype, alpha thalassemia produces a significant decrease in erythrocyte potassium chloride co-transporter (KCC). This may be at least one way in which the presence of alpha thalassemia leads to a milder phenotype in sickle-cell disease, since KCC is markedly upregulated in sickle RBCs (especially in HbSC sickle-cell disease) and is believed to play an important role in RBC dehydration. Indeed, in HbSC sickle-cell disease, a correlation has been found between KCC activity and disease severity. However, alpha thalassemia was not found to impact survival in patients with HbSS, although “a nonsignificant trend was seen in those with HbSC.” [36]
Patient Education
Patients with a family history or a known carrier state for alpha thalassemia gene mutations should obtain genetic counseling to determine their genotype and assess the risk to offspring. This is also true in cases of suspected concomitant hemoglobinopathy.
-
Alpha-chain genes in duplication on chromosome 16 pairing with non-α chains to produce various normal hemoglobins.
-
The severity of alpha thalassemia is primarily dependent on the number of α-globin genes that have been deleted or inactivated, but it can be affected by co-existing factors as well.
-
Distribution of α+ thalassemia (orange) and α0 thalassemia (yellow).
-
This demonstrates α0 thalassemia resulting from deletions that remove large portions of DNA on the same allele (“in cis”), including, in some cases, the embryonic zeta-globin gene (eg, --THAI, --FIL).
-
Peripheral smear from patient with hemoglobin H (HbH) disease showing target cells, microcytosis, hypochromia, and anisopoikilocytosis. Morphologic abnormalities are similar to those observed in beta thalassemia. In silent carriers, only mild microcytosis is observed.
