Overview
Surprisingly, many upper extremity malformations cause little functional deficit. [1] Children develop prehension with hands as they are, and they usually are not self-conscious of difference until they become socialized in school. In contrast, parents may be dismayed by the appearance of an anomalous hand and may be hoping that surgery can creat a "normal" hand. The hand surgeon treating children with upper extremity anomalies must offer surgery to improve the child's function and cosmesis, when possible, and counsel parents about what is and is not possible with surgery.
Timing of surgery
Early surgery is defined as that performed within the first 2 years of life. Advantages include the full potential for growth, development, and patterns of use; improved scarring; early incorporation of the reconstructed part; and reduced psychologic affect. Disadvantages of early surgery are increased technical difficulty and possible increased anesthetic risk. Most surgeons perform these operations when the patient is in the second year of life but no later than when the patient enters school.
Incidence
The best epidemiologic studies of the incidence of congenital anomalies are total population studies. A 5-year Edinburgh birth registry study by Rogala et al found the prevalence of babies born with any limb anomalies to be 30 cases per 10,000 live births and the incidence of upper limb anomalies to be 22.5 cases per 10,000 live births. [2] Of those with upper limb anomalies, 35% had another non–upper limb anomaly. They used an older classification, so some direct comparisons to more recent studies are difficult.
An 11-year total population study of Western Australia found the prevalence of babies born with upper limb anomalies to be 19.76 cases per 10,000 live births. [3] Forty-six percent of those affected had another nonhand congenital anomaly. Fifty-two percent had bilateral hand anomalies, and 17% had multiple different hand anomalies. No significant differences in prevalence or frequency of anomalies were found between whites and people of color, left and right sides, and in babies who survived and those who died shortly after birth. [3]
Similarly, an 11-year total population study of the Stockholm region of Sweden found a recorded incidence of congenital anomalies of the upper limb of 21.5 cases per 10,000 live births. [4] Fifty-four percent of the children with congenital anomalies of the upper limb were boys. The anomalies affected the right side only in 30%, the left side only in 33%, and both sides in 37%. Nonhand anomalies were recorded in 23% of the children with congenital anomalies of the upper limb, most commonly in the lower limbs. In 17% of the effected children, there was a known occurrence among relatives. [4]
These population studies are slightly higher than the estimated 0.16-0.18% incidence by extrapolations of patients presenting for treatment, [1, 5, 6] as it is thought that milder deformities may never present for treatment.
A study by Goldfarb et al of congenital upper limb anomalies in a group of Midwestern US patients found that of 480 extremities with a malformation, 62% had anomalies of the hand plate alone, with radial polydactyly (15%), symbrachydactyly (13%), and cleft hand (11%) being the most common of these. [7]
Embryology
Limb development takes place during the third to eighth week of gestation. The anteroposterior (AP, or preaxial-postaxial, or radial-ulnar) axis is the first to be established. Before the limb bud is present, there is specialized mesoderm in what will become the posterior aspect of the limb bud called the zone of polarizing activity (ZPA). The ZPA secretes a protein, Sonic Hedgehog (SHH), which regulates the AP axis. The ectoderm in the dorsum of the developing limb bud secretes a protein called wingless-type mouse mammary tumor virus integration site 7a (Wnt-7a). Wnt-7a stimulates secretion of transcription factor Lmx1b, which causes dorsal development in this part of the limb bud. In the ventral limb bud, the transcription factor engrailed-1 (En-1) is produced. En-1 blocks Wnt-7a expression, preventing expression of Lmx-1b there, and establishes the dorsal-ventral axis. [8, 9]
SHH also plays a role in establishing the proximal-distal (PD) axis of the limb bud. It stimulates the apical ectodermal ridge (AER), the ectoderm at the tip of the developing limb bud, to secrete several different fibroblast growth factors (FGF), especially FGF-2, FGF-4, and FGF-8. These regulate the proximal-distal development of the limb bud. When stimulation of the AER by SHH halts, the distal development of the limb bud also halts. Wnt-7a also stimulates SHH production, indirectly influencing the AP and PD axes of development. [8, 9]
Table 1. Three Spatial Axes in Limb Embryology [10, 11] (also see the diagrams below Table 1) (Open Table in a new window)
Signaling Center |
Signals involved |
Action |
AER |
FGFs (FGF-8 distally) |
Proximal-to-distal orientation |
ZPA |
SHH: GLi3A (posterior)/Gli3R (anterior) |
Radial-to-ulnar orientation |
Ectoderm |
WNT pathway: Wnt7a & Lmx-1b (dorsal)/engrailed-1 (ventral) |
Dorsal-to-ventral orientation |
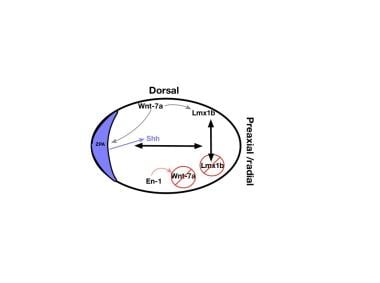 Diagrammatic cross-section of the limb bud. The dorsal ectoderm produces wingless-type mouse mammary tumor virus integration site 7a (Wnt-7a), which stimulates production of transcription factor Lmx1b. In the ventral ectoderm, engrailed 1 gene product (En1) blocks production of Wnt-7a and, in turn, Lmx1b. The gradient of Lmx1b establishes dorsal-ventral differentiation. Wnt-7a also stimulates zone of polarizing activity (ZPA) production of Sonic hedgehog protein (SHH).
Diagrammatic cross-section of the limb bud. The dorsal ectoderm produces wingless-type mouse mammary tumor virus integration site 7a (Wnt-7a), which stimulates production of transcription factor Lmx1b. In the ventral ectoderm, engrailed 1 gene product (En1) blocks production of Wnt-7a and, in turn, Lmx1b. The gradient of Lmx1b establishes dorsal-ventral differentiation. Wnt-7a also stimulates zone of polarizing activity (ZPA) production of Sonic hedgehog protein (SHH).
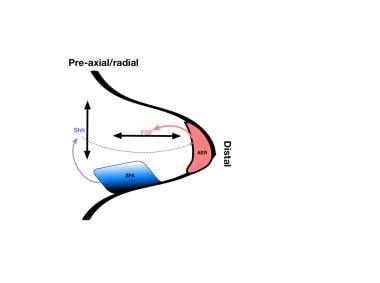 Diagrammatic limb bud with factors responsible for axes of development. The zone of polarizing activity (ZPA) produces Sonic Hedgehog protein (SHH), which controls the radial/ulnar axis. SHH also stimulates the apical ectodermal ridge (AER) to produce fibroblast growth factors (FGFs), which control the proximal-distal axis.
Diagrammatic limb bud with factors responsible for axes of development. The zone of polarizing activity (ZPA) produces Sonic Hedgehog protein (SHH), which controls the radial/ulnar axis. SHH also stimulates the apical ectodermal ridge (AER) to produce fibroblast growth factors (FGFs), which control the proximal-distal axis.
The hand plate develops in the fifth week of gestation. Homeobox transcription factors (HOX) and SHH together determine digit number and identity. SHH induces an ulnar-to-radial gradient of bone morphogenic proteins (BMPs) that induces programmed cell death (apoptosis) in the interdigital space by suppressing the FGF expression in the overlying AER. BMP also helps establish digital identity by maintaining FGF in the AER overlying digits and inducing sex-determining region Y-related, high-mobility group box 9 (SOX9) there. SOX9 regulates chondrogenesis in the phalanx-forming (PFR) region of the developing hand. It is not clear how the family of BMPs interacts to produce the pattern of alternating digit development and web space recession. [8, 12] See the diagrams below.
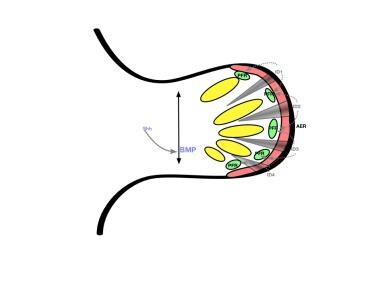 Early hand formation: Sonic Hedgehog protein (SHH) stimulation results in anteroposterior bone morphogenic protein gradient in the handplate. This gradient causes formation of interdigital (ID) signalling centers, which stimulate the phalanx-forming region (PFR) and repress the interdigital apical ectodermal ridge (AER).
Early hand formation: Sonic Hedgehog protein (SHH) stimulation results in anteroposterior bone morphogenic protein gradient in the handplate. This gradient causes formation of interdigital (ID) signalling centers, which stimulate the phalanx-forming region (PFR) and repress the interdigital apical ectodermal ridge (AER).
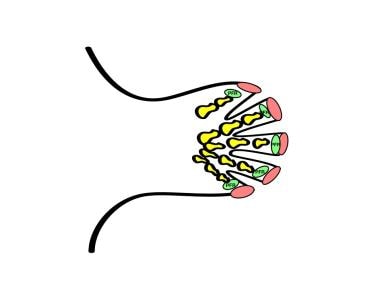 Repression of the interdigital apical ectodermal ridge (AER) effects apoptosis of the interdigital region. In the digit, the AER stimulates outgrowth, and the phalanx-forming region (PFR) regulates digital development.
Repression of the interdigital apical ectodermal ridge (AER) effects apoptosis of the interdigital region. In the digit, the AER stimulates outgrowth, and the phalanx-forming region (PFR) regulates digital development.
Vessels and then nerves subsequently grow into the limb. Mesodermal differentiation into cartilage and muscle begins to occur. The limb begins to pronate, the elbow flexes, and the hand is flexed and ulnarly deviated. Ossification of the phalanges takes place antenatally, while the carpal bones ossify postnatally. By the third week, vessels begin to penetrate the mesodermal mass. Myogenic regions of the limb become vascular, and chondrogenic regions become avascular. Large interosseous and median arteries later give way to the radial and ulnar arteries.
Muscle formation is derived from a dorsal (extensor) and ventral (flexor) muscle blastoma. Tendons develop independently of their muscle bellies, only linking later in development. If the adjacent bone is not developed, the tendon attaches to the nearest adjacent structure. It is unknown whether the specificity of the nerve-muscle relationship is random or specific. Mixed motor and sensory nerves enter the limb as a pioneer growth cone. In general, the anterior division of these nerves supplies the flexors and the posterior division supplies the extensors.
Dysmorphology
The causes of congenital anomalies can be divided conveniently into genetic, environmental, and unknown causes. A minority of congenital anomalies have a single major environmental or genetic cause.
Genetic causes of congenital anomalies are gradually becoming elucidated; for instance, GLI3 gene expression in the developing limb bud is modulated by the SHH protein gradient (See the diagrams below).
Diagrammatic cross-section of the limb bud. The dorsal ectoderm produces wingless-type mouse mammary tumor virus integration site 7a (Wnt-7a), which stimulates production of transcription factor Lmx1b. In the ventral ectoderm, engrailed 1 gene product (En1) blocks production of Wnt-7a and, in turn, Lmx1b. The gradient of Lmx1b establishes dorsal-ventral differentiation. Wnt-7a also stimulates zone of polarizing activity (ZPA) production of Sonic hedgehog protein (SHH).
Diagrammatic limb bud with factors responsible for axes of development. The zone of polarizing activity (ZPA) produces Sonic Hedgehog protein (SHH), which controls the radial/ulnar axis. SHH also stimulates the apical ectodermal ridge (AER) to produce fibroblast growth factors (FGFs), which control the proximal-distal axis.
With complete absence of GLI3, the hand develops without radial-to-ulnar digit differentiation, instead developing with an increased number of identical "default" digits. In the usual clinical presentation, GLI3 may be more locally effected, yielding less complete expression. In the absence of some of the HOX genes, the interdigital space or PFR are disrupted, causing polydactyly, syndactyly, or polysyndactyly. [12]
Multiple problems in morphogenesis may be explained in terms of a sequence, which is a simple problem leading to a cascade of subsequent problems. Developmentalists have designated the following 3 types of sequences:
-
Malformation sequence, in which an intrinsic malformation exists in the embryo, resulting in certain other abnormalities (eg, radial dysplasia)
-
Deformation sequence, in which no intrinsic defect is found in the embryo; rather, an abnormal external mechanical or structural force results in secondary distortion or deformation (eg, constriction bands)
-
Disruption sequence, in which the healthy embryo is subjected to tissue breakdown or injury (TORCH [ie, toxoplasmosis, other infections, rubella, cytomegalovirus infection, and herpes simplex] group is in this category, as are thalidomide-caused deformities)
When the cause is unknown, the term malformation is preferred. Approximately one half of cases with multiple anomalies fall into known syndromes.
Genetics
Although at least 112 recognized syndromes are described with hand anomalies as a part of their expression, this represents only 5% of congenital hand anomalies. [1]
As time goes on, more congenital upper limb anomalies are being matched to gene loci and genes. See Table 2 below.
Table 2. Known Gene Loci and Gene Associations With Congenital Upper Limb Anomalies (Open Table in a new window)
Classification |
Syndrome |
Locus |
Gene |
Polydactyly |
|
|
|
Preaxial |
|
|
|
Isolated |
Preaxial polydactyly II/triphalangeal thumb |
7q36 |
C7orf2/ZRS |
|
Preaxial polydactyly IV |
7p13 |
GLI3 |
Associated |
Orofacial digital syndrome II |
|
|
|
Townes-Brocks |
16q12.1 |
SALL1 |
Postaxial |
|
|
|
Isolated |
Postaxial polydactyly A1 |
7p13 |
GLI3 |
|
Postaxial polydactyly A2 |
13q21-q32 |
|
|
Postaxial polydactyly A3 |
19p13.1 |
|
|
Postaxial polydactyly B |
7p13 |
GLI3 |
Associated |
Ellis-van Creveld |
4p16 |
EVC |
|
Smith-Lemili-Opitz |
11q12 |
DHCR7 |
|
McCusick-Kaufmann |
20q12 |
BB56 |
|
Bardet-Biedl 1 |
11q13 |
|
|
Bardet-Biedl 2 |
16q21 |
|
|
Bardet-Biedl 3 |
3p13 |
|
|
Bardet-Biedl 4 |
15q22 |
|
|
Bardet-Biedl 5 |
2q31 |
|
|
Bardet-Biedl 7 |
4q27 |
|
|
Meckel-Gruber |
17q21 |
|
|
Jeune |
15q13 |
|
Central |
Pallister-Hall |
7p13 |
|
Preaxial and Postaxial |
Greig |
7p13 |
|
|
|
|
|
Syndactyly |
|
|
|
Isolated |
Syndactyly 1 |
2q34-q36 |
|
|
Syndactyly 2 (synpolydactyly) |
2q31-q32 |
HOXD13 |
|
Syndactyly 3 (oculodentodigital) |
6q22-q24 |
CX43 |
Associated |
Apert |
10q26 |
FGFR2 |
|
Pfeiffer |
8p11.2 |
FGFR1 |
|
|
10q26 |
FGFR2 |
|
Fraser |
4q21 |
FRAS1 |
|
|
|
|
Brachydactyly |
|
|
|
Isolated |
Brachydactyly A1 |
2q35 |
IHH |
|
Brachydactyly A2 |
4q23-q24 |
BMPR18 |
|
Brachydactyly C |
9q34 |
ROR2 |
|
Brachydactyly B |
20q11.2 |
CDMP1 |
|
Brachydactyl D |
2q31 |
HOXD13 |
|
Brachydactyly E |
2q31 |
HOXD13 |
Associated |
Robinow |
9q34 |
ROR2 |
|
Grebe |
20q11.2 |
CDMP1 |
|
Rubenstein-Taybi |
16p13.3 |
CREBBP |
|
Turner |
45XO |
|
|
Albright hereditary Osteodystrophy |
20q13.2 |
GNAS |
|
Gorlin basal cell nevus syndrome |
9q22.3 |
PTC |
|
Bilginturan hypertension with Brachydactyly |
12p |
|
|
|
|
|
Reduction |
|
|
|
Mesoaxial |
|
|
|
Isolated |
Split hand-foot malformation 1 |
7q21.3-q22.1 |
DLX5/6 |
|
Split hand-foot malformation 2 |
Xq26 |
|
|
Split hand-foot malformation 3 |
10q24-35 |
|
|
Split hand-foot malformation 4 |
3q27 |
P63 |
|
Split hand-foot malformation 5 |
2q31 |
|
|
Acheiropodia |
7q36 |
C7orf2 |
Associated |
Ectrodactyly ectodermal Dysplasia |
3q27 |
PG6 |
|
Holt-Oram |
12q24.1 |
TBX5 |
|
Hand-foot-genital |
7p14.2-p15 |
HOXA13 |
Postaxial |
Cornelia de Lange |
3q26 |
|
|
Ulna mammary |
12q24.1 |
TBX3 |
Classification
The classification proposed by Swanson has been adopted by the American Society for Surgery of the Hand (ASSH) and the International Federation of Societies for Surgery of the Hand (IFSSH). [13]
Type I - Failure of formation, as follows:
See the list below:
-
Transverse arrest - Can be at any level, shoulder to phalanx
-
Longitudinal arrest - Can include preaxial (varying degrees of hypoplasia of the thumb or radius); central (divided into typical and atypical types of cleft hand); postaxial (varying degrees of ulnar hypoplasia to hypothenar hypoplasia); and intercalated longitudinal arrest (various types of phocomelia)
Type II - Failure of differentiation, as follows:
See the list below:
-
Soft tissue - Syndactyly, trigger thumb, Poland syndrome, camptodactyly
-
Skeletal - Various synostoses and carpal coalitions
-
Tumorous conditions - Include all vascular and neurologic malformations
Type III - Duplication, as follows:
See the list below:
-
May apply to whole limb, mirror hand, polydactyly
Type IV - Overgrowth, as follows:
See the list below:
-
Includes conditions such as hemihypertrophy and macrodactyly
Type V - Undergrowth, as follows:
See the list below:
-
Most commonly, radial hypoplasia, brachysyndactyly, or brachydactyly
Type VI - Constriction band syndromes, as follows:
See the list below:
-
Occurs with or without distal lymphedema; may involve amputation at any level
Type VII - Generalized anomalies and syndromes
This classification system is imperfect (eg, atypical cleft hands are difficult to classify, falling into 1 of 3 possible groups), and the future undoubtedly holds a classification system based on improved knowledge of the molecular genetics underlying the anomalies. [14, 9]
The modification of the IFSSH system of Manske and Oberg [15] incorporates recent advances in understanding embryology and shares the IFSSH system's advantage of simplicity of use. [12]
Group I. Failure of axis formation or differentiation, as follows:
See the list below:
-
Radial longitudinal deficiency
-
Radial-ulnar synostosis
-
Ulnar longitudinal deficiency
-
Transverse deficiency (including symbrachydactyly)
-
Dorsal ventral deficiency
Group II. Failure of hand-plate formation and/or differentiation, as follows:
See the list below:
-
Syndactyly
-
Apert Syndrome
-
Central deficiency (cleft hand)
-
Camptylodactyly
-
Clinodactyly
-
Clasped Thumb
-
Hand-plate synostosis - Metacarpal synostosis, carpal synostosis
Group III. Duplication, as follows:
See the list below:
-
Radial Polydactyly (including triphalangeal thumb)
-
Ulnar Dimelia
-
Ulnar Polydactyly
Group IV. Overgrowth
Group V. Amniotic band sequence
Group VI. Generalized skeletal abnormalities
Time will tell if this system gains the widespread adaption similar to the system of Swanson.
Type I - Failure of Formation
Transverse arrest at the arm level may be difficult to distinguish from the proximal constriction band syndromes although these are usually bilateral. Treatment is by prosthesis.
Intercalated phocomelia
In amelia, no hand structure is present. In phocomelia, a functional terminal element is always present. The types of phocomelia are as follows:
-
Type 1 - those in which the hand attaches to the shoulder (forearm and arm deficient)
-
Type 2 - those in which the forearm attaches to the shoulder (arm deficient)
-
Type 3 - those in which the hand attaches to the arm (forearm deficient)
These patients are often best treated by the prosthetist. When the phocomelic hand can reach the mouth, even this is not indicated. Newer myoelectric prostheses allow muscle action to trigger the prosthesis. Occasionally, radial or ulnar deviation of the hand may require centralization.
Transverse arrest at forearm level
The forearm is the most common level for transverse arrest. Transverse arrest may be bilateral, in contrast with constriction ring syndrome, which is usually not. Treatment of these patients is by prosthetics. Prosthetic fitting with a mitten is now recommended in patients starting at age 6 months to allow the child to get accustomed to the use of a prosthesis. This avoids substitution patterns. A hook can be added when the patient is aged 2 years. Children with unilateral deformities tend to discard the prosthesis despite encouragement. The most accepted prosthesis is a simple cosmetic arm, not the more functional myoelectric.
Transverse arrest at the carpal, metacarpal, and phalangeal level
Transverse arrest at this level is classified as acheiria (ie, absence of hand) or adactyly (ie, absence of fingers). These are often difficult to distinguish from constriction band syndromes or as part of a cleft hand. A nail remnant may be present in constriction band syndrome but not in transverse arrest. When a mobile wrist is present, a palmar plate prosthesis is a good treatment option. If metacarpal elements are present, functional potential is increased greatly, and the objective should be the creation of a basic hand.
Microvascular transfer of multiple toes is method for reconstruction of a functional hand in selected patients. [16, 17] Transfer of free nonvascularized toe phalanges is another established method of reconstruction for adactyly. [18, 19] Recent studies have shown significant persistent donor deficits with this technique, however. [20, 21] Bourke and Kay proposed reconstruction of donor toes with iliac crest bone graft. [20]
Preaxial deficiency
These deficiencies may vary from minor abnormalities of the thenar muscles to complete absence of the preaxial structures. The most common presentation is the short radius, absence of scaphoid and trapezium, and total absence or hypoplasia of the thumb (ie, radial club hand). Elbow stiffness may follow ulnar dislocation due to growth restriction by the fibrous anlage. It occurs bilaterally as frequently as unilaterally. In the former, it is part of a syndrome in 77% of patients; it is part of a syndrome in 40% of patients when unilateral. [22]
Syndromes associated with preaxial deficiency include the following:
-
Holt-Oram syndrome - Heart defects, most commonly septal defects
-
Thrombocytopenia-absent radius (TAR) syndrome - Thrombocytopenia present at birth but improves with time
-
VACTERL - Vertebral abnormalities, anal atresia, cardiac abnormalities, tracheoesophageal fistula, esophageal atresia, renal defects, radial dysplasia, lower limb abnormalities
-
Fanconi anemia - Aplastic anemia not present at birth; develops around 6 years of age
Fanconi anemia is fatal without a bone marrow transplant; therefore, all patients with radial deficiency should be tested for Fanconi anemia. [23]
The thumb may be absent or hypoplastic (see Type V - Undergrowth). Digital stiffness may be observed, heralded by the lack of extension creases. O'Rahilly classified this anomaly as follows, according to the development of the radius [24] :
-
Type I - Short radius, no radial bowing or deviation, no treatment
-
Type II - Hypoplastic radius rare, no treatment
-
Type III - Partial absence with fibrous anlage most common; requires centralization procedure
-
Type IV - Total absence second most common; elbow joint usually deficient; soft tissue release and centralization
James et al classified these on a broader array of findings, as shown in Table 3 below. [25]
Table 3. Thumb Anomaly Classification (Open Table in a new window)
Type |
Thumb Anomaly |
Carpal Anomaly |
Distal Radius |
Proximal Radius |
N |
Absence or hypoplasia |
Normal |
Normal |
Normal |
0 |
Absence or hypoplasia |
Absence, hypoplasia, or coalition |
Normal |
Normal, synostosis, or radial head dislocation |
1 |
Absence or hypoplasia |
Absence, hypoplasia, or coalition |
>2 mm shorter than ulna |
Normal, synostosis, or radial head dislocation |
2 |
Absence or hypoplasia |
Absence, hypoplasia, or coalition |
Hypoplasia |
Hypoplasia |
3 |
Absence or hypoplasia |
Absence, hypoplasia, or coalition |
Absence of physis |
Hypoplasia |
4 |
Absence or hypoplasia |
Absence, hypoplasia, or coalition |
Absence |
Absence |
The radial nerve often ends at the elbow, with the median nerve substituting distally. The radial artery is usually absent. The anlage may restrict growth on the radial side such that ulnar dislocation occurs at the elbow.
Treatment of preaxial deficiencies
O'Rahilly types I and II often do not require treatment. Some patients should not undergo surgical repair, including those with inadequate elbow flexion, in which straightening the wrist may make it impossible for the patient to bring the hand to the mouth [26] .
Splints, casts, and passive stretching are useful to maintain soft tissue length. Surgical management has the following 4 elements:
-
Soft tissue release: This is performed prior to the centralization and involves an aggressive capsular release.
-
Centralization or radialization of the carpus: This technique is usually best when the patient is younger than 3 years but only in patients in whom the elbow has a good range of motion (ROM). The fibrous anlage is excised, and the wrist is fixed with a central K wire.
-
Thumb reconstruction, often by pollicization of the index finger (see Type V - Undergrowth)
Central deficiency
This deficiency manifests as the absence of digits or metacarpals within the central portion of the hand. The radius and ulna are normal. Barsky classified them as being either typical or atypical. [27] Miura and Suzuki summarized the differences between the 2 as shown below in Table 4. [28]
Table 4. Typical vs Atypical Cleft Hand (Open Table in a new window)
|
Typical Cleft Hand |
Atypical Cleft Hand |
Clinical features |
||
Involvement |
Bilateral |
Unilateral |
Inheritance |
Familial |
Spontaneous |
Syndactyly |
Common |
Rare |
Poland Syndrome association |
No |
Yes |
Cleft lip and palate association |
Yes |
No |
Anatomic findings |
||
Arterial supply |
Ring finger may have 3 arteries |
Vestigial supply to central digits |
Tendon |
Dual tendons to ring finger |
Minimal |
Skeleton |
Hypertrophy adjacent to cleft |
Hypoplasia |
Classification |
Failure of differentiation |
Failure of formation |
Postaxial deficiencies
Postaxial deficiencies are characterized by a much greater degree of variation than those on the preaxial side. They are much rarer, with only 1 postaxial deficiency per every 10 preaxial deficiencies. Most show autosomal dominant (AD) patterns of inheritance. Syndromic associations, by contrast, are rare. Postaxial deficiencies usually are unilateral compared to preaxial deficiencies, which are 100% bilateral. Bayne's classification (as follows) is similar to O'Rahilly's classification of radial deficiency: [29]
-
Type I - Deficient ulna, minimal deformity
-
Type II - Partial absence with fibrous anlage radial bowing
-
Type III - Total absence with anlage; radius may be straight
-
Type IV - Humeroradial synostosis; entire limb is shortened
Cole and Manske developed a classification based on the development of the hand, as follows: [30]
-
Type A - Normal first web space and normal thumb
-
Type B - Mild deficiency of first web space and mild thumb hypoplasia with intact opposition and extrinsic tendon function
-
Type C - Moderate-to-severe deficiency of first web space and similar thumb hypoplasia with loss of opposition and dysfunction of extrinsic tendons
-
Type D - Absence of thumb
Partial absence is the most common variety (ie, Bayne type II). Carpal coalitions and thumb anomalies are also common. The extent of the hand anomaly may bear no correlation with the forearm defect. Unlike preaxial anlages, excision probably is warranted only if ulnar deviation is present at the wrist. Radial head dislocation may limit elbow function. Function at the wrist is generally good, and the ulnar nerve is usually present.
Treatment of postaxial deficiencies
Therapy and splinting are used to improve passive range of motion. Resection of the anlage is controversial, since ulnar deviation is never severe, the ulnar tilt of the radial physis is usually minimal, radial head dislocation is not progressive, and the functional results without resection are excellent. Creation of a 1-bone forearm by fusion of the proximal ulna to the distal radius solves the problem of radial head dislocation and ulnar bowing. This may create a stable forearm with a better appearance.
Type II - Failure of Differentiation
Syndactyly
Syndactyly is one of the most common congenital anomalies in the hand. Failure of programmed cell death in the seventh week of gestation results in this anomaly. The exception is acrosyndactyly, in which refusion of distal digits occurs as part of the constriction ring syndrome. The incidence varies with race but is approximately 1 in 2000. It is usually bilateral, and males are affected more commonly than females. Many different types of inheritance patterns are documented (being a common association). The third and then fourth web in the hand and the second web in the foot are most affected. Syndactyly is associated with at least 28 syndromes. [31]
Syndactyly is classified according to completeness (complete, incomplete) and the presence of bony union (simple, complex, complicated). Complicated syndactyly refers to any syndactyly in which more than simple side-side bony fusion exists. Associated growth abnormalities with buckling of the bones are common. The normal web ends midway down the proximal phalanx. The skin demonstrates an hourglass configuration, with a 45° slope dorsal to palmar. The fingers are joined by fascial connections across the syndactyly, which link with Cleland ligaments. These are responsible for growth retardation. Phalangeal malformations are common, such as the middle phalanx in clinodactyly and the delta proximal phalanx in the thumb. Symphalangism is common, particularly in syndromic syndactyly.
Treatment
The goals of surgery are separation of the digits, provision of a lined commissure, and avoidance of scars. Innumerable techniques exist but most embody the following principles:
-
Either a dorsal or volar flap [32]
-
Zigzag incisions to lessen the contracture
-
Operating on one side of the digit at a time
-
Correction of skeletal abnormality
-
Creation of a normal nail
-
Full-thickness skin graft (FTSG) for defects to avoid contracture
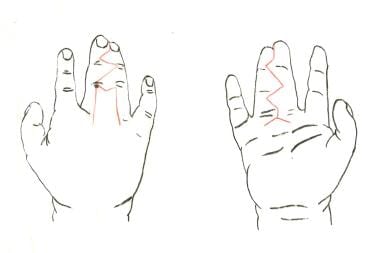 Complete simple syndactyly of middle and ring fingers. Incisions are planned in red.
Complete simple syndactyly of middle and ring fingers. Incisions are planned in red.
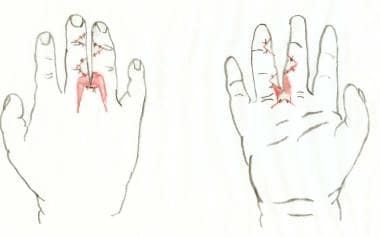 After division of syndactyly, with flaps inset. Skin grafts (pink) are needed proximally.
After division of syndactyly, with flaps inset. Skin grafts (pink) are needed proximally.


The dorsal flap is raised, taking care to preserve the dorsal vein. Neurovascular bundles are identified and preserved, and the fascial bands connecting to Cleland ligaments are divided. The proximal division of the common digital nerve is often the structure limiting the proximal release. The transverse metacarpal ligament rarely may need to be excised. Excess defatting of the flaps to secure closure should be avoided. The site of skin graft harvest should not be a future hair-bearing area. Techniques that use all of the skin for one finger and graft the other generally yield poor results. [33] Leave one digit visible to enable positional checking within the cast.
First web syndactyly occurs in less than 10% of syndactyly incidents. Usually, a single large Z-plasty or a 4-flap Z-plasty is sufficient. The investing fascia of the thenar muscles and the adductor pollicis may require release. In these situations, a skin graft is necessary, with K wire fixation to maintain the intraoperative gains.
In a single-surgeon study, Mericli et al reported that the use of a tapered M-to-V flap was an effective means of web space reconstruction in the management of syndactyly. The study, which included 89 primary congenital hand and 32 foot syndactylies, reported a complication rate of 14%, with no total flap losses. The investigators claimed that M-to-V flaps have advantages over rectangular flaps, including easier intraoperative adjustment and, through Z-plasty at the palmodigital crease, minimization of scar contracture. [34]
Wang et al described a syndactyly repair technique not requiring skin grafts; instead, they used a dorsal island flap advanced into the web space. [35] Wang and Hutchinson performed a study of 14 patients treated for bilateral syndactyly with a standard technique employing both skin flaps and grafts on one side, and skin flaps only on the other. They found that, while tourniquet time was longer on the grafted side, that side had less web creep and superior cosmesis. [36]
Indeed, since the turn of the 21st century, there has been a trend in syndactyly surgery toward flap use alone, with the avoidance of skin grafts. This is considered advantageous since the transplanted skin can, owing to an inconsistent growth rate, lead to scarring and contracture. The flap/nongraft surgery is indicated in simple, incomplete syndactyly that extends no further than the proximal interphalangeal joint. Extensive defatting is required so that the surface area of the separated fingers is not “too great for the skin to rectify.” If the defatting is too extreme, however, patients may experience neurovascular damage, venous drainage problems, and an emaciated appearance to the fingers. [37]
Timing
Patterns of prehension are established by the time the individual is aged 1 year. Therefore, all severe abnormalities should be corrected by this time. Correction of less-severe deformities can be deferred to age 12-36 months. Bilateral procedures are best performed early, prior to the time the patient is aged 2-3 years.
Complications
These can include the following [38] :
-
Vascular compromise - Due to tight skin closure or tight dressings
-
Infection or maceration
-
Web creep - Correlated strongly with wound sepsis. Also may reflect a growth discrepancy between the scar tissue and surrounding normal tissue. Interdigital splints of limited value [39]
-
Hypertrophic scarring
A study by McQuillan et al found a low 30-day acute complication rate for corrective syndactyly and polydactyly surgery. Using the American College of Surgeons National Surgical Quality Improvement Program database, the investigators examined information for four different Current Procedural Terminology codes, including for simple syndactyly, complex syndactyly, and polydactyly operations. Of 1656 cases of congenital hand anomalies, the report found an overall complication rate of 2.2%, with superficial surgical site infection being the most frequent complication (1.7%). Patients who underwent surgery for complex syndactyly had a higher complication rate (5.2%) than did the other patients (2.3%). [40]
Chouairi et al found evidence that ventilator dependence, tracheostomy, and preoperative comorbidities are significantly more prevalent among individuals with complex, rather than simple, syndactyly. The investigators included 956 patients in the study, with the cohort reflecting patterns in the operative treatment of syndactyly in the United States. [41]
Syndactyly-Related Syndromes
Apert syndrome
Apert syndrome is an acrocephalosyndactyly syndrome (APS). Several other APS exist, but they tend to have less severe hand deformities. Apert syndrome is a result of a mutation of fibroblast growth factor receptor type 2 gene, on chromosome 10q, and inheritance is autosomal dominant. [23] The craniofacial deformity consists of bicoronal synostosis and midface hypoplasia; the hands show symmetrical complex syndactyly. Dysplasia of the shoulder and elbow joint are also common. The hand deformity fits into 3 general patterns, as outlined in Table 5 below. [42]
Table 5. Classification of Apert Hand Deformity (Open Table in a new window)
Classification |
1st Web Space |
Central Mass |
4th Web Space |
Type I: Spade hand |
Incomplete simple syndactyly |
Digital mass flat to palmar plane |
Incomplete simple syndactyly |
Type II: Mitten or spoon hand |
Complete simple syndactyly |
Digital mass curved due to fingertip fusion |
Complete simple syndactyly |
Type III: Hoof or rosebud hand |
Complete complex syndactyly |
Thumb incorporated into mass, synechia of nails |
Simple syndactyly, metacarpal fusion |
Common to all types of hand are a short radially deviated thumb, complex syndactyly, symphalangism of all 4 digits, and a simple syndactyly of the fourth web.
Correction of the craniofacial deformity usually is done first. Correction of hand deformities is recommended when the patient is younger than 2 years. The principles of treatment are to lengthen and realign the thumb and to separate the digits, beginning with the border digits. Resection of the index finger may be necessary to create a reasonable first web space. The resultant 3-fingered hand may be an aesthetic and functional improvement. In the central hand, releasing only one side at a time is advisable. The thumb may be corrected by a wedge osteotomy and lengthened as necessary. The little (L) finger is often the most normal finger in the hand.
Poland syndrome
Poland syndrome is likely the result of disruption of blood flow in the embryonic subclavian artery. Unilateral absence of the sternocostal head of the pectoralis major is the most constant deficit, but the pectoralis minor, latissimus dorsi, serratus, and deltoid muscles also may be hypoplastic. Rib cage deficiencies, scoliosis, and dextrocardia have been described. The hand deformity consists of brachydactyly, hand hypoplasia, and simple syndactyly. The central digits are most often affected.
The treatment of syndactyly in Poland syndrome usually is as described for simple syndactyly.
Symphalangism
The term symphalangism was introduced by Cushing and describes the failure of segmentation that results in stiff fingers. This is usually at the proximal interphalangeal (PIP) joint, with metacarpophalangeal (MP) fusion rare. The 3 types of symphalangism are as follows:
-
In true symphalangism, the digits are of normal length.
-
In brachysymphalangism, the digits are short as well as stiff.
-
Syndromic symphalangism may be related to specific syndromes, particularly Poland and Apert syndromes.
The joint is represented by a solid cartilaginous bar, which appears as a joint on radiographs. Affected digits are more slender and lack the flexion creases, which is the clue to making the diagnosis.
Attempts at surgical joint reconstruction have met with poor results. Arthrodesis or osteotomy may be useful after the patient reaches maturity. Recommended angles are 20°/30°/40°/50° for the respective digits.
"Thumb in palm" deformities
Infants normally hold their thumbs in flexion until aged 3 months, possibly due to the dominance of the flexor innervation prior to that time. Lack of extension (ie, clasped thumb posture) at this time may be due to trigger thumb, congenital absence of the thumb extensors, camptodactyly, or arthrogryposis.
Flexion deformities secondary to extensor hypoplasia may be isolated to one finger or involve all fingers. The thumb is involved most commonly, and when the thumb is involved, the index (I) and middle (M) fingers also may be involved. Metaphalangeal (MP) extension is lacking, while the interphalangeal (IP) joints can be extended by the intrinsics.
Passive ROM must be maintained by passive ROM exercises. If an established contracture exists, then joint release is necessary to ensure full ROM prior to tendon transfer. Two options are as follows:
Camptodactyly
Camptodactyly has been divided into the following 3 categories [43] :
-
Type I - Congenital, isolated unilateral, usually only the small finger. This type affects males and females equally.
-
Type II - Similar to type I, but becomes apparent between ages 7-11 y; affects females much more often than males. This is a progressive flexion deformity that does not resolve spontaneously.
-
Type III - Severe, congenital, effecting multiple digits bilaterally. This type is associated with a variety of generalized conditions, as follows:
Arthrogryposis
Craniofacial disorders
Orofacial digital syndrome
Craniocarpotarsal dystrophy (Freeman-Sheldon syndrome)
Oculodentodigital dysplasia
Oculopalatodigital syndrome
Chromosomal disorders
Trisomy 13-15
Short stature
Camptomelic dysplasia
Mucopolysaccharidosis
Facial-digital-genital (Aaskorg-Scott syndrome)
Others
Osteo-onychodysostosis (Turner-Kieser syndrome)
Cerebrohepatorenal (Zellweger syndrome)
Virtually every structure in the vicinity of the proximal interphalangeal (PIP) joint has been implicated in the pathogenesis of camptodactyly. Most quote a dynamic imbalance caused by abnormal intrinsic anatomy, with bony and capsular changes secondary. This is most likely to be either an aberrant lumbrical insertion into the flexor digitorum superficialis (FDS) or the digital sheath or an aberrant or tight FDS origin. Other theories are a tight FDS tendon or a palmar plate contracture. Radiographs show a flattening of the condyle of the head of the proximal phalanx. The bony changes are believed to be the result of, not the cause of, contractures.
Initial treatment should be conservative by splinting and stretching, although splinting may be difficult to maintain. Only one half of adolescent patients improve with stretching. Patients with severe contractures and those in whom conservative management fails are surgical candidates. In general, the principles are as follows:
-
Identify the anatomic variant.
-
Release an anomalous lumbrical or lengthen a tight FDS.
-
If the PIP can be extended actively with the wrist and MPs flexed, then FDS release alone may be indicated.
-
Permanent correction may require tendon transfer. The two slips of FDS can be transferred to the extensor retinaculum.
In long-standing contractures, the optimal result may be achieved only by arthrodesis in the optimal position or by osteotomy.
Clinodactyly
Clinodactyly denotes a deviation of a finger as a result of an abnormally shaped middle phalanx. Inward deviation of the L finger is the most common presentation and occurs in as many as 20% of non-Caucasian individuals. The second most common presentation is the proximal phalanx of the thumb. It is almost ubiquitous in widespread congenital anomalies, hence the term "background noise." The degree of deformity correlates directly with the number of associated anomalies. Up to 80% of children with Down syndrome have clinodactyly.
Three treatment options are available, namely opening wedge osteotomy with nonvascularized bone graft (NVBG), closing wedge osteotomy, or reversed wedge osteotomy. A further option is the excision of bone and replacement by a free fat graft. A closing wedge osteotomy may result in a mallet finger deformity.
Delta phalanx
This also is termed congenital triangular bone and may refer to a metacarpal. The classic finding is a bone with a continuous C-shaped physis extending the length of the shortened side, like a staple. It usually involves either the middle phalanx of the L finger or the proximal phalanx of the thumb (eg, Apert syndrome). It is part of the deformity in triphalangeal thumb. The clinical presentation thus is clinodactyly but it is due to the delta phalanx. In infancy, it merely can be excised. Later in life, an osteotomy or arthrodesis may be necessary. Excision of the longitudinal physis with fat interposition has been suggested. In the thumb, an opening wedge osteotomy and NVBG, centralization of the flexor and extensor tendons, and reattachment of the intrinsics may be indicated.
Radioulnar synostosis
Radioulnar synostosis occurs more commonly than generally is accepted. Longitudinal segmentation of the radius and ulna proceeds from distal to proximal; failure to differentiate results in synostosis. Sixty percent of cases of radioulnar synostosis affect only a single arm on the patient. This condition presents as a fixed pronation deformity. The radial head is either absent and involved in the synostosis or dislocated. Differentiating this condition from posttraumatic synostosis is important.
Treatment is indicated only for bilateral synostosis or for bilateral synostosis in which the fixed pronation deformity is greater than 45°. Most authors are pessimistic about the results of operative treatment. The treatment of choice is a derotational osteotomy through the synostosis. The major risks are vascular compromise and posterior interosseous nerve palsy. Previously, one arm was placed in slight supination and the other in pronation. Most now recommend neutral and 20° pronation, respectively.
Humeroradial synostosis
The joint is represented by a solid cartilaginous bar, which appears as a joint on radiographs. Affected digits are more slender and lack the flexion creases, which is the clue to making the diagnosis.
Attempts at surgical joint reconstruction have met with poor results. Arthrodesis or osteotomy may be useful after the patient reaches maturity. Recommended angles are 20°/30°/40°/50° for the respective digits.
Type III - Duplication
Polydactyly
Polydactyly is the most common congenital anomaly of the upper limb. [44] In England, it was believed to relate to royalty. Total duplication of the hand and forearm is ulnar dimelia (rare). Central duplication is relatively rare, although it often is reported as syndactyly, thus underestimating its incidence. It probably results from increased folding of the AER.
Central duplication may be termed polysyndactyly. Complex central reduplications require individualized treatment. Conversion to a 3-finger hand may be the best option in some patients. [45]
Isolated postaxial polydactyly has a strong autosomal dominant inheritance pattern and is common in individuals of African descent. [46]
Preaxial polydactyly (i.e. duplicated thumb) has been classified by Wassel (see image below). [47]
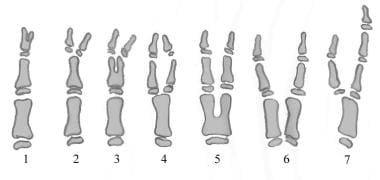 Diagram of Flatt (Wassel) classification of thumb duplication (radial polydactyly) in s-rays of skeletally immature hand. Class 7 is a triphalangeal thumb with or without duplication at any level.
Diagram of Flatt (Wassel) classification of thumb duplication (radial polydactyly) in s-rays of skeletally immature hand. Class 7 is a triphalangeal thumb with or without duplication at any level.
A Mexican study, by Ortiz-Cruz et al, found that in isolated postaxial polydactyly, male prevalence (6.35/10,000) was significantly greater than female (5.45/10,000). Hands were affected more often than feet, and left limbs more frequently than right ones, with incomplete extradigit formation more prevalent than complete formation (74.50% vs 25.50%, respectively). [48]
Ikuta reported the relative incidence of 267 cases of thumb polydactyly as identified by the Wassel classification (see Table 6 below). [49]
Table 6. Thumb Polydactyly Incidence (Open Table in a new window)
Wassel Class |
Incidence, % |
1 |
5.3 |
2 |
20.1 |
3 |
4.6 |
4 |
31.9 |
5 |
3.9 |
6 |
8.2 |
7 |
13.8 |
Surgical correction of complex duplications is timed according to most other anomalies, as a compromise between delay on technical grounds and early operation for functional reasons. For example, treatment of thumb polydactyly must take into consideration that development of pinch occurs in the first year of life. The principle of surgical treatment is to ablate the most useless digit and to reconstruct its partner. The collateral ligaments of joints must be reconstructed. The most common complication is persistence or recurrence of angulation. [50]
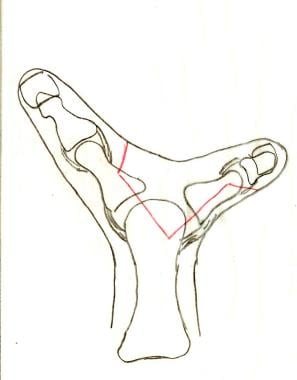 Flatt (Wassel) type 4 duplication of the thumb. Incisions are planned in red; the digit to the left is to be preserved.
Flatt (Wassel) type 4 duplication of the thumb. Incisions are planned in red; the digit to the left is to be preserved.
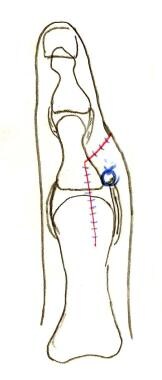 The preserved digit is centralized by repairing the collateral ligament from the ablated digit (blue suture).
The preserved digit is centralized by repairing the collateral ligament from the ablated digit (blue suture).
In distal thumb duplication (Wassel classes 1 and 2), Bilhaut-Cloquet advocated removing the central triangular portion of the distal phalanx and nail, leaving a midline scar. [51] Reports of scarring and separation of the nail have made this method less satisfactory. [49] Baek et al, however, have demonstrated very good results using a modified Bilhaut-Cloquet technuique. [52]
A study by de Almeida of patients surgically treated for duplicated thumbs, including 18 patients who underwent removal of the radial digit, found that the best results could be obtained when surgery is carried out prior to age 3 years, with all anatomic differences addressed (especially in Wassel class 7) in order to decrease the likelihood of residual deformity. [53]
Mirror hand (ulnar dimelia)
The classic presentation of ulnar dimelia is duplication of the ulna, with no radius, and 7 or 8 fingers with no thumbs. It may occur in association with fibular dimelia and absence of the tibia. Ulnar dimelia may represent an additional polarizing zone within the limb bud. The postaxial digits usually are more functional. The index finger may not be duplicated (thus 7 fingers) or may be syndactylized to its partner. The fingers are named index, middle, ring, small, and accessory index, middle, ring, and small. Elbow motion is usually limited, with the elbow normally held in extension. [54]
In the Entin procedure, the accessory preaxial middle and small fingers are resected, and the accessory ring digit is used as a thumb by a modified pollicization procedure. An alternate approach is to use the index finger as part of a conventional pollicization, if this appears to be a more functional digit. No tissue should be discarded until it clearly is not necessary for the reconstruction. Wrist extension is restored by tendon transfer (finger extensors from discarded fingers to become wrist extensors). [54]
Type IV - Overgrowth
Macrodactyly
This condition should be differentiated from others in which an enlargement may occur (ie, vascular malformations, enchondromatoses, osteoid osteoma, fibrous dysplasia, lipomatoses). Macrodactyly is nevertheless a heterogenous group, with 4 of the common presentations as follows:
-
Macrodactyly associated with nerve-orientated lipofibromatosis: This is the most common form with no inheritance pattern. It alternately is described as nerve-territory orientated macrodactyly (NTOM). It most commonly occurs in the median nerve distribution. Two types of growth occur: static, in which the growth is proportional to that of the child; and progressive, in which it is more rapid. The outstanding feature is the fatty infiltration of neural tissue. Tsuge believes the overgrowth to be "nerve-driven" (nerve resection sometimes cures it).
-
Macrodactyly associated with neurofibromatosis: Gigantism of the upper or lower limbs is a well-known concomitant of neurofibromatosis. Diagnostic criteria are as follows:
6 cafe au lait spots greater than 2.5 cm
Multiple neurofibromas along peripheral nerves
Pedunculated cutaneous tumors termed molluscum fibrosum
-
It is similar in presentation to macrodactyly associated with nerve-orientated lipofibromatosis but also may involve osteocartilaginous outgrowths. Unlike in the lipofibromatosis-associated type, digital enlargement is seldom unilateral. No marked fatty infiltration of peripheral nerves is present, although the same degree of perineural fibrosis is present.
-
Macrodactyly associated with hyperostosis: In this form, no neural abnormality occurs. Osteochondral masses, but not enchondromata, are present.
-
Macrodactyly/gigantism and hemihypertrophy: This is rare.
The progressive type of macrodactyly is particularly debilitating, functionally and psychologically. Individualization of treatment is particularly important, perhaps more so than in other forms of congenital hand surgery. The objectives are to diminish length and bulk without compromising sensation or vascularity. Surgical options include the following:
-
Epiphysiodesis or total physeal resection
-
Staged debulking (can incorporate longitudinal bony resection and excision or osteochondral masses)
-
Nerve stripping or resection and end-to-end coaptation
-
Shortening procedures: In Tsuge's version, a dorsal dog-ear results; in the Barsky version, the dog-ear is on the ventral surface
-
Partial amputation or ray amputation
Type V - Undergrowth
Hypoplasia of the thumb is frequently associated with radial dysplasia (see Type I - Failure of Formation). Blauth classified hypoplasia of the thumb, as shown below in Table 7. [55]
Table 7. Hypoplasia of the Thumb (Open Table in a new window)
Type |
Findings |
Treatment |
1 |
Minimal shortening |
None or augmentation |
2 |
Narrow first web space, thenar weakness, MCP joint instability |
Opponensplasty, release of first web space, UCL reconstruction |
3 |
Narrow first web space, thenar weakness, MCP joint instability, extrinsic tendon abnormalities |
|
3A |
Metacarpal hypoplasia with stable CMC joint |
Reconstruction |
3B |
Metacarpal hypoplasia with unstable CMC joint |
Pollicization |
4 |
Rudimentary phalanges, thumb attached by a skin bridge |
Pollicization |
5 |
Aplasia of thumb |
Pollicization |
The critical determinant of the reconstructive plan is the condition of the basilar joint. If the basilar joint is stable, reconstructing the existing thumb is worthwhile; if the basilar joint is unstable, removing the thumb and performing a pollicization is usually best. [56] Littler and Buck-Gramko described the technique of pollicization as follows: [57]
Skin incisions are designed to allow proximal transposition and pronation of the index finger into the thumb's position. The shaft of the index metacarpal is resected, preserving the metacarpal head, which will function as the new trapezium. The index is isolated as a neuromuscular island so that it may be moved to the new more proximal position. The index must be rotated into pronation to be positioned to oppose the other fingers. The metacarpal head should be rotated into flexion to prevent the new thumb CMC joint from hyperextending as the index MCP joint does. The extrinsic tendons need to be shortened to function effectively, and the intrinsic muscles are reattached. First dorsal interosseous is sutured into the radial lateral band, to function as the new abductor pollicis brevis; the first palmar interosseous is sutured into the ulnar lateral band to function as the adductor pollicis.
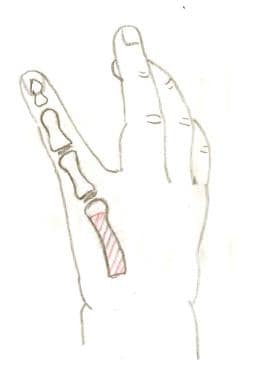 A right hand with aplasia of the thumb. The shaft of the index metacarpal will be removed for pollicization.
A right hand with aplasia of the thumb. The shaft of the index metacarpal will be removed for pollicization.
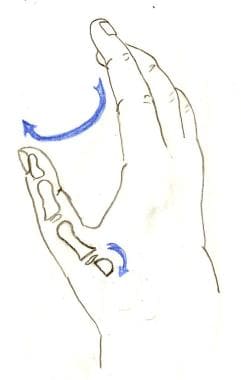 The index finger is pronated in order to oppose the other digits (large blue arrow). The metacarpal head is rotated into flexion in order to prevent hyperextension (small blue arrow).
The index finger is pronated in order to oppose the other digits (large blue arrow). The metacarpal head is rotated into flexion in order to prevent hyperextension (small blue arrow).
Other surgical options for thumb reconstruction, if the basilar joint is deemed stable, include the following:
-
First web space deepening
-
Metacarpal transposition from the center to the border of the hand
-
Skeletal distraction methods [58]
-
Use of an incomplete index finger as a neurovascular island flap to lengthen the thumb, known as the on-top plasty (vascularized bone graft [VBG])
-
Nonvascularized phalangeal transfer from the foot; in the phalangeal transfers (nonvascularized), 90% growth has been recorded when transferred with periosteum
-
Nonvascularized bone graft
-
Vascularized toe-to-thumb transfers [56]
Type VI - Constriction Band Syndromes
This syndrome represents a disruption or deformation sequence subsequent to annular bands of chorionic tissue encircling the limb. It has a low incidence of associated anomalies, normal proximal anatomy, and a sporadic occurrence. Clubfoot is the most common association. Distal fusion of amputated digits, as in acrosyndactyly, may be present. Patterson described specific diagnostic criteria as follows: [59]
-
Simple constriction ring
-
Constriction ring with distal deformity (lymphedema)
-
Constriction ring with distal soft tissue fusion
-
Intrauterine amputations
Treatment is individualized according to the deformity. Options are as follows:
-
Excision of the groove with multiple Z-plasty or W-plasty
-
Excision of functionless nubbins of tissue
-
Pollicization for patients in whom the thumb is absent
Acrosyndactyly
Acrosyndactyly is considered to be part of the constriction band syndrome. [60] The band may be present at the birth, and histologic studies have shown this to be chorion. The literal translation means digits fused together in a peak. It supposedly is due to a disruption sequence after the digits have separated, in which they are rejoined by scar tissue. The pattern of limb involvement is completely random. Proximal epithelial-lined sinuses are present and lie more distal than normal commissures, with distal amputation common. Because of the precarious nature of the blood supply, several smaller surgical procedures are preferred to one larger one.
Type VII - Generalized Anomalies and Syndromes
Arthrogryposis multiplex congenita
This is a syndrome of unknown etiology that presents at birth and manifests with multiple joint contractures. A decrease in the number of anterior horn cells is observed, with degenerative changes within striated muscles. The joint contractures are probably secondary. Classification into myopathic or neuropathic types is made on the basis of muscle biopsy. Intelligence is normal. It is a diagnosis of exclusion, with marked joint contractures the cardinal sign. The skin is atrophic and waxy in appearance. The hand usually is clasped with a thumb-in-palm deformity.
Shoulder and elbow ROM must be established by therapy and splinting prior to attending to the hand. The principles of management are to release joint contractures and to provide appropriate motors for the joints by judicious tendon transfers.
Vascular malformations
See Vascular, Capillary Malformations; Vascular, Hemangiomas; Vascular, Lymphatic Malformations; and Vascular, Venous Malformations.
-
Diagrammatic cross-section of the limb bud. The dorsal ectoderm produces wingless-type mouse mammary tumor virus integration site 7a (Wnt-7a), which stimulates production of transcription factor Lmx1b. In the ventral ectoderm, engrailed 1 gene product (En1) blocks production of Wnt-7a and, in turn, Lmx1b. The gradient of Lmx1b establishes dorsal-ventral differentiation. Wnt-7a also stimulates zone of polarizing activity (ZPA) production of Sonic hedgehog protein (SHH).
-
Diagrammatic limb bud with factors responsible for axes of development. The zone of polarizing activity (ZPA) produces Sonic Hedgehog protein (SHH), which controls the radial/ulnar axis. SHH also stimulates the apical ectodermal ridge (AER) to produce fibroblast growth factors (FGFs), which control the proximal-distal axis.
-
Early hand formation: Sonic Hedgehog protein (SHH) stimulation results in anteroposterior bone morphogenic protein gradient in the handplate. This gradient causes formation of interdigital (ID) signalling centers, which stimulate the phalanx-forming region (PFR) and repress the interdigital apical ectodermal ridge (AER).
-
Repression of the interdigital apical ectodermal ridge (AER) effects apoptosis of the interdigital region. In the digit, the AER stimulates outgrowth, and the phalanx-forming region (PFR) regulates digital development.
-
Complete simple syndactyly of middle and ring fingers. Incisions are planned in red.
-
After division of syndactyly, with flaps inset. Skin grafts (pink) are needed proximally.
-
Diagram of Flatt (Wassel) classification of thumb duplication (radial polydactyly) in s-rays of skeletally immature hand. Class 7 is a triphalangeal thumb with or without duplication at any level.
-
Flatt (Wassel) type 4 duplication of the thumb. Incisions are planned in red; the digit to the left is to be preserved.
-
The preserved digit is centralized by repairing the collateral ligament from the ablated digit (blue suture).
-
A right hand with aplasia of the thumb. The shaft of the index metacarpal will be removed for pollicization.
-
The index finger is pronated in order to oppose the other digits (large blue arrow). The metacarpal head is rotated into flexion in order to prevent hyperextension (small blue arrow).
Tables
- Table 1. Three Spatial Axes in Limb Embryology [10, 11] (also see the diagrams below Table 1)
- Table 2. Known Gene Loci and Gene Associations With Congenital Upper Limb Anomalies
- Table 3. Thumb Anomaly Classification
- Table 4. Typical vs Atypical Cleft Hand
- Table 5. Classification of Apert Hand Deformity
- Table 6. Thumb Polydactyly Incidence
- Table 7. Hypoplasia of the Thumb
Signaling Center |
Signals involved |
Action |
AER |
FGFs (FGF-8 distally) |
Proximal-to-distal orientation |
ZPA |
SHH: GLi3A (posterior)/Gli3R (anterior) |
Radial-to-ulnar orientation |
Ectoderm |
WNT pathway: Wnt7a & Lmx-1b (dorsal)/engrailed-1 (ventral) |
Dorsal-to-ventral orientation |
Classification |
Syndrome |
Locus |
Gene |
Polydactyly |
|
|
|
Preaxial |
|
|
|
Isolated |
Preaxial polydactyly II/triphalangeal thumb |
7q36 |
C7orf2/ZRS |
|
Preaxial polydactyly IV |
7p13 |
GLI3 |
Associated |
Orofacial digital syndrome II |
|
|
|
Townes-Brocks |
16q12.1 |
SALL1 |
Postaxial |
|
|
|
Isolated |
Postaxial polydactyly A1 |
7p13 |
GLI3 |
|
Postaxial polydactyly A2 |
13q21-q32 |
|
|
Postaxial polydactyly A3 |
19p13.1 |
|
|
Postaxial polydactyly B |
7p13 |
GLI3 |
Associated |
Ellis-van Creveld |
4p16 |
EVC |
|
Smith-Lemili-Opitz |
11q12 |
DHCR7 |
|
McCusick-Kaufmann |
20q12 |
BB56 |
|
Bardet-Biedl 1 |
11q13 |
|
|
Bardet-Biedl 2 |
16q21 |
|
|
Bardet-Biedl 3 |
3p13 |
|
|
Bardet-Biedl 4 |
15q22 |
|
|
Bardet-Biedl 5 |
2q31 |
|
|
Bardet-Biedl 7 |
4q27 |
|
|
Meckel-Gruber |
17q21 |
|
|
Jeune |
15q13 |
|
Central |
Pallister-Hall |
7p13 |
|
Preaxial and Postaxial |
Greig |
7p13 |
|
|
|
|
|
Syndactyly |
|
|
|
Isolated |
Syndactyly 1 |
2q34-q36 |
|
|
Syndactyly 2 (synpolydactyly) |
2q31-q32 |
HOXD13 |
|
Syndactyly 3 (oculodentodigital) |
6q22-q24 |
CX43 |
Associated |
Apert |
10q26 |
FGFR2 |
|
Pfeiffer |
8p11.2 |
FGFR1 |
|
|
10q26 |
FGFR2 |
|
Fraser |
4q21 |
FRAS1 |
|
|
|
|
Brachydactyly |
|
|
|
Isolated |
Brachydactyly A1 |
2q35 |
IHH |
|
Brachydactyly A2 |
4q23-q24 |
BMPR18 |
|
Brachydactyly C |
9q34 |
ROR2 |
|
Brachydactyly B |
20q11.2 |
CDMP1 |
|
Brachydactyl D |
2q31 |
HOXD13 |
|
Brachydactyly E |
2q31 |
HOXD13 |
Associated |
Robinow |
9q34 |
ROR2 |
|
Grebe |
20q11.2 |
CDMP1 |
|
Rubenstein-Taybi |
16p13.3 |
CREBBP |
|
Turner |
45XO |
|
|
Albright hereditary Osteodystrophy |
20q13.2 |
GNAS |
|
Gorlin basal cell nevus syndrome |
9q22.3 |
PTC |
|
Bilginturan hypertension with Brachydactyly |
12p |
|
|
|
|
|
Reduction |
|
|
|
Mesoaxial |
|
|
|
Isolated |
Split hand-foot malformation 1 |
7q21.3-q22.1 |
DLX5/6 |
|
Split hand-foot malformation 2 |
Xq26 |
|
|
Split hand-foot malformation 3 |
10q24-35 |
|
|
Split hand-foot malformation 4 |
3q27 |
P63 |
|
Split hand-foot malformation 5 |
2q31 |
|
|
Acheiropodia |
7q36 |
C7orf2 |
Associated |
Ectrodactyly ectodermal Dysplasia |
3q27 |
PG6 |
|
Holt-Oram |
12q24.1 |
TBX5 |
|
Hand-foot-genital |
7p14.2-p15 |
HOXA13 |
Postaxial |
Cornelia de Lange |
3q26 |
|
|
Ulna mammary |
12q24.1 |
TBX3 |
Type |
Thumb Anomaly |
Carpal Anomaly |
Distal Radius |
Proximal Radius |
N |
Absence or hypoplasia |
Normal |
Normal |
Normal |
0 |
Absence or hypoplasia |
Absence, hypoplasia, or coalition |
Normal |
Normal, synostosis, or radial head dislocation |
1 |
Absence or hypoplasia |
Absence, hypoplasia, or coalition |
>2 mm shorter than ulna |
Normal, synostosis, or radial head dislocation |
2 |
Absence or hypoplasia |
Absence, hypoplasia, or coalition |
Hypoplasia |
Hypoplasia |
3 |
Absence or hypoplasia |
Absence, hypoplasia, or coalition |
Absence of physis |
Hypoplasia |
4 |
Absence or hypoplasia |
Absence, hypoplasia, or coalition |
Absence |
Absence |
|
Typical Cleft Hand |
Atypical Cleft Hand |
Clinical features |
||
Involvement |
Bilateral |
Unilateral |
Inheritance |
Familial |
Spontaneous |
Syndactyly |
Common |
Rare |
Poland Syndrome association |
No |
Yes |
Cleft lip and palate association |
Yes |
No |
Anatomic findings |
||
Arterial supply |
Ring finger may have 3 arteries |
Vestigial supply to central digits |
Tendon |
Dual tendons to ring finger |
Minimal |
Skeleton |
Hypertrophy adjacent to cleft |
Hypoplasia |
Classification |
Failure of differentiation |
Failure of formation |
Classification |
1st Web Space |
Central Mass |
4th Web Space |
Type I: Spade hand |
Incomplete simple syndactyly |
Digital mass flat to palmar plane |
Incomplete simple syndactyly |
Type II: Mitten or spoon hand |
Complete simple syndactyly |
Digital mass curved due to fingertip fusion |
Complete simple syndactyly |
Type III: Hoof or rosebud hand |
Complete complex syndactyly |
Thumb incorporated into mass, synechia of nails |
Simple syndactyly, metacarpal fusion |
Wassel Class |
Incidence, % |
1 |
5.3 |
2 |
20.1 |
3 |
4.6 |
4 |
31.9 |
5 |
3.9 |
6 |
8.2 |
7 |
13.8 |
Type |
Findings |
Treatment |
1 |
Minimal shortening |
None or augmentation |
2 |
Narrow first web space, thenar weakness, MCP joint instability |
Opponensplasty, release of first web space, UCL reconstruction |
3 |
Narrow first web space, thenar weakness, MCP joint instability, extrinsic tendon abnormalities |
|
3A |
Metacarpal hypoplasia with stable CMC joint |
Reconstruction |
3B |
Metacarpal hypoplasia with unstable CMC joint |
Pollicization |
4 |
Rudimentary phalanges, thumb attached by a skin bridge |
Pollicization |
5 |
Aplasia of thumb |
Pollicization |

What would you like to print?
- New Treatment Recommendations for Hand Osteoarthritis
- Crinecerfont Promising for Congenital Adrenal Hyperplasia
- Drug Bests Phototherapy for Severe Hand Eczema
-
 PIK3CA-Related Overgrowth Spectrum (PROS): 5 Things to Know
PIK3CA-Related Overgrowth Spectrum (PROS): 5 Things to Know
- Understanding the Causes and Repercussions of Delayed Presentation of Congenital Hand Anomalies
- A New Understanding and a Minimalist Approach for Rhinoplasty


- Overview
- Incidence
- Embryology
- Dysmorphology
- Genetics
- Classification
- Type I - Failure of Formation
- Type II - Failure of Differentiation
- Type III - Duplication
- Type IV - Overgrowth
- Type V - Undergrowth
- Type VI - Constriction Band Syndromes
- Type VII - Generalized Anomalies and Syndromes
- Show All
- Media Gallery
- Tables
- References