Background
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/ARVC) is an inherited cardiomyopathy characterized by structural and functional abnormalities in the right ventricle (RV) resulting in ventricular arrhythmias. [1] This primary disease of heart muscle leads to fibrofatty replacement of the RV and the subepicardial region of the left ventricle. It is an important cause of sudden cardiac death (SCD) in young adults, accounting for 11% of all cases and 22% of cases among athletes. [2, 3] (See the image below.)
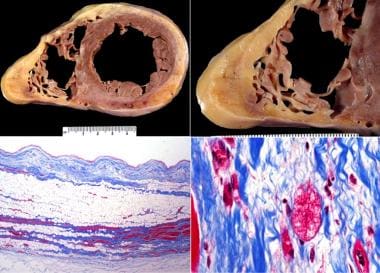 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). ARVD/ARVC, biventricular, autopsy heart in a young man who died suddenly playing basketball. Top left demonstrates increased fat in the outer walls of the right ventricle and left ventricular posterolateral walls. A higher magnification of the right ventricle is seen at the top right image; the anterior wall is nearly completely replaced with fat, and fibrofatty irregular posterior wall involvement is seen. Note that no actual thinning of the wall itself exists, although the muscular portion is in some areas completely missing. The bottom left image demonstrates a full thickness of the right ventricle stained with Masson trichrome. The residual muscle is present only in a bandlike area of scarring, and subepicardial scarring is present as well. The characteristic myocyte vacuolization, depicted in the bottom right image, is seen in nearly all areas of ARVD/ARVC within the scarred areas.
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). ARVD/ARVC, biventricular, autopsy heart in a young man who died suddenly playing basketball. Top left demonstrates increased fat in the outer walls of the right ventricle and left ventricular posterolateral walls. A higher magnification of the right ventricle is seen at the top right image; the anterior wall is nearly completely replaced with fat, and fibrofatty irregular posterior wall involvement is seen. Note that no actual thinning of the wall itself exists, although the muscular portion is in some areas completely missing. The bottom left image demonstrates a full thickness of the right ventricle stained with Masson trichrome. The residual muscle is present only in a bandlike area of scarring, and subepicardial scarring is present as well. The characteristic myocyte vacuolization, depicted in the bottom right image, is seen in nearly all areas of ARVD/ARVC within the scarred areas.
ARVD was first described in 1977 and was included in the World Health Organization (WHO) classification of cardiomyopathies in 1996. [4] Since then, there have been significant advances in the understanding of its etiopathogenesis, diagnosis, and management. [5]
Pathophysiology
The structural abnormalities in arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/ARVC) result from the fatty infiltration and fibrosis of the RV myocardium. This leads to progressive RV dilatation and dysfunction. The left ventricle (LV) is less commonly involved, and the septum is relatively spared. [6] However, in a cohort of 200 probands, Sen-Chowdhry et al found that LV involvement may even precede the onset of significant RV dysfunction. [7] The prognosis is worse in patients with LV involvement. [8]
The mechanisms for myocardial loss include the following:
-
Apoptosis (programmed cell death)
-
Inflammation, enhanced fibrosis, and loss of function
-
Fatty replacement of myocardium
Etiology
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/ARVC) is an inherited disorder, as it is already present in the fetus. Familial cases account for 30-90% of cases. [9] In other cases, it may result from an acquired etiology such as viral infection (myocarditis) or unidentified inheritance. It is also likely that patients with a genetic predisposition are more likely to develop myocarditis.
The disease manifests more frequently in active individuals, when mechanical sheer stress can cause cell membrane damage, inflammation, and fibrosis in genetically predisposed RV.
Genetics
ARVD is considered a genetic disorder, as most cases are familial, and there is geographical clustering. The most common pattern of inheritance is autosomal dominance, with a variable penetrance ranging from 20-35% of family members. [10, 11] People living in the Veneto region of Italy have a higher penetrance. The autosomal-recessive (Naxos disease) pattern of inheritance is localized to the Greek island of Naxos and is associated with palmoplantar keratosis and wooly hair. The genetic mutation occurs on chromosome 17q21, and penetrance is almost 100%. [12, 13]
Genetic abnormalities in ARVD are located on chromosomes 1, 2, 3, 6, 7, 10, 12, and 14. There is no single unique genetic abnormality, posing a challenge in evaluation of patients and families with suspected ARVD. The responsible genes include plakoglobin (JUP), desmoplakin (DSP), plakophilin-2 (PKP2), desmoglein-2 (DSG2), desmocollin-2 (DSC2), and others. [14, 15, 16] In some cases, mutation in the SCN5A gene may cause dysfunction in the cardiac voltage-gated sodium channel (Nav1.5), resulting in cardiomyopathy. [17]
The Heart Rhythm Society and the European Heart Rhythm Association published a consensus statement on genetic testing for cardiomyopathies. [18]
Epidemiology
Because of the diagnostic challenge, the exact incidence and prevalence of arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/ARVC) remains unknown, as clinically silent cases may go unrecognized. [19] It is estimated that it affects 1 in 2000 to 1 in 5000 in the general population [19, 20] and is more common in individuals of Greek and Italian origin. [21]
In a cohort of 100 patients from the United States, the median age at presentation was 26 years, and 51% were males. The median time to diagnosis was one year from the initial presentation, and median survival in the entire cohort was 60 years. [22]
Prognosis
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/ARVC) is an important cause of sudden cardiac death in young adults, accounting for 11% of all cases and 22% of cases among athletes.
The prognosis is worse in patients with left ventricular (LV) involvement.
Pregnancy is mostly well tolerated, but Hodes et al found 13% of pregnancies in women with ARVD/ARVC were complicated by ventricular arrhythmias and 5% by heart failure. [23]
Sustained ventricular arrhythmias are more common in men, and they have significantly abnormal electrocardiograms. [24]
Cardiac magnetic resonance imaging (CMRI) to assess tissue characteristics and a 5-year risk score model has been proposed. [25] In a study of 140 patients with ARVC, CMRI was normal in 14 patients who did not have any major events. LV-dominant abnormalities were found in 16 patients (12%). [26] Isolated RV involvement was seen in 41% patients and biventricular involvement was seen in 37% patients. LV involvement had a worse prognosis than RV involvement alone. However, the risk score model underestimated the risk in those who had LV involvement. [26]
-
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). Intraventricular conduction abnormality.
-
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). T-wave abnormalities in the presence of right bundle branch block.
-
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). Anteroseptal T-wave inversion.
-
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). Apical four-chamber transthoracic echocardiogram (TTE) showing right ventricular dilatation and dysfunction.
-
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). Parasternal long-axis view with right ventricular outflow tract dilatation and dysfunction.
-
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). Axial black blood image of the right ventricle showing fatty infiltration in the free wall of the right ventricle.
-
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). Arrhythmogenic right ventricular dysplasia epsilon wave.
-
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). ARVD/ARVC, biventricular, autopsy heart in a young man who died suddenly playing basketball. Top left demonstrates increased fat in the outer walls of the right ventricle and left ventricular posterolateral walls. A higher magnification of the right ventricle is seen at the top right image; the anterior wall is nearly completely replaced with fat, and fibrofatty irregular posterior wall involvement is seen. Note that no actual thinning of the wall itself exists, although the muscular portion is in some areas completely missing. The bottom left image demonstrates a full thickness of the right ventricle stained with Masson trichrome. The residual muscle is present only in a bandlike area of scarring, and subepicardial scarring is present as well. The characteristic myocyte vacuolization, depicted in the bottom right image, is seen in nearly all areas of ARVD/ARVC within the scarred areas.
-
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). Gross findings at autopsy, ARVD/ARVC. A suspicion of the diagnosis occurs if the right ventricle appears dilated, especially fatty, upon initial inspection of the heart (top left). Actually, aneurysms occur in approximately 35% of patients when evaluated by imaging. At autopsy, dilatation such as seen in this example occurs in 40-50% of cases, with true aneurysm being less common. The fibrofatty involvement of the right ventricle (upper right) is typically patchy; in this case, a segment of fatty replacement exists, without significant thinning or aneurysm, features that were once considered pathognomonic of the disease. Left ventricular scarring is generally subepicardial, but in this case (bottom left), the scarring is more random, reflecting the phenotypic heterogeneity of ARVD/ARVC. On cross-section (short-axis cuts) seen at low magnification (bottom right), the changes are not particularly striking of a specific cardiomyopathy; it is almost the rule that careful examination for fibrofatty infiltrates is necessary for adequate sampling that will lead to the diagnosis.
-
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC). The top left demonstrates an area of fat in the right ventricle, with fibrous tissue highlighted by the Masson trichrome stain. The areas of fat are irregular, in contrast to the normal "marbling" appearance seen in the anterior right ventricle, in which relatively even, linear fatty areas exist. The fat in ARVD/ARVC is considered "metaplastic" or secondary to an area of previous myocyte destruction. In contrast to the right ventricle, the areas of involvement in the left ventricle (top right) often show fat and more fibrous tissue. A higher magnification demonstrates altered myocytes with vacuoles, which are seen in all areas of fibrofatty infiltration (bottom left). Depending on the degree of sampling and definition of myocarditis, inflammation with myocyte necrosis is seen in over 25% of cases (bottom right).
