Practice Essentials
Choroid plexus papilloma (CPP) is a benign but rare central nervous system (CNS) neoplasm of the choroid plexus—a structure made from tufts of villi within the ventricular system that produces cerebrospinal fluid (CSF). [1, 2, 3] Choroid plexus papillomas may be seen at any age but most commonly arise in children younger than 5 years. [4] A vast majority of these neoplasms are benign, but a small percentage are malignant. [5, 6, 7, 8, 9, 10] Children usually present with increasing head circumference or altered mental status, whereas adults present with signs of increased intracranial hypertension. Imaging shows intraventricular enhancing masses. [11]
Choroid plexus papillomas account for about 1% of all brain tumors, 2-4% of brain tumors in children younger than 15 years, 10-20% of brain tumors that occur in the first year of life, and 0.5% of adult brain tumors. [11, 12] The most common location is the atrium of the lateral ventricle in children and the fourth ventricle in adults. Rare locations include the third ventricle, the cerebellopontine angle (CPA), and the cerebral parenchyma. [8]
Symptoms seen with choroid plexus tumors generally result from blocking of the CSF pathways and debatably by secretion of CSF by tumor cells, both leading to increased fluid and, eventually, to hydrocephalus. Not infrequently, the tumor itself can cause mass effect, with symptoms dependent on tumor location. In either case, eventual progression and increased intracranial pressure can occur. Cases of hydrocephalus occasionally are not resolved with surgery, possibly because of derangement of reabsorption mechanisms or blockage at other sites in the ventricular system.
Symptoms of food refusal, apathy, speechlessness, and low mood have been reported. [12] Patients with CPP typically present with signs and symptoms of hydrocephalus. Some CPPs are diagnosed prenatally, but many of them reach a large size before diagnosis. These neoplasms have traditionally been treated with surgical resection. Treatment presents a challenge due to risk of high blood loss during resection. [13] Additional therapeutic options may be necessary in cases of remnant tumor portions, recurrence, or malignant transformation. [14]
Patients usually present with the following signs of increased intracranial pressure: headache, nausea and vomiting, drowsiness, ocular or gaze palsies (cranial nerves [CN] III and VI), papilledema, visual disturbances, and, eventually, blindness. Infants, especially those with a tumor located in the third ventricle, can present with hydrocephalus or macrocephalus, as well as with associated increased intracranial pressure. Unusual presentations include trochlear palsies (CN IV), psychosis, or, occasionally, seizures.
As CPPs grow, they eventually obstruct the flow of CSF. Once the intracranial space can no longer compensate for the increase in pressure, a tension-obstruction type of hydrocephalus develops. Persistently increased intracranial pressure is not compatible with life. Pressure is alleviated by tumor resection or by a ventricular shunting procedure.
As with most CNS neoplasms, confinement to the intracranial cavity is usual; however, CPPs may seed cells into the CSF leading to rare drop metastases. If myelopathic symptoms are present, consideration of dissemination into the spinal canal should warrant spinal cord neuroimaging studies.
Contraindications to surgical correction of CPP are based on the patient's comorbidities and on his or her ability to tolerate surgery. However, watchful waiting is inappropriate in most cases. As choroid plexus tumors grow, resulting hydrocephalus and other complications usually lead to greater morbidity than that which occurs if tumors are removed when they are first discovered and are smaller.
(An image depicting a choroid plexus papilloma can be seen below.)
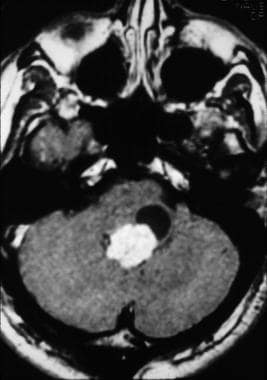
SEER data
The Surveillance and End Results (SEER) database was reviewed by Cannon et al for population-based outcomes of choroid plexus tumors (CPTs), including choroid plexus papillomas (CPPs), atypical CPPs (aCPPs), and choroid plexus carcinomas (CPCs). A total of 349 patients with CPT were identified (120 CPCs, 26 aCPPs, and 203 CPPs). Patients with CPC presented at a younger age (median, 3 yr; mean, 14.8 yr) relative to those presenting with CPP (median, 25 yr; mean, 28.4 yr). Histology was a significant predictor of overall survival (OS), with 5-year OS rates of 90% for CPP, 77% for aCPP, and 58% for CPC. Older age and male sex were prognostic for worse OS and cause-specific survival for CPP. Only extent of surgery had a significant impact on survival for CPC. [15]
In another review of SEER data, by Dudley et al, of 107 CPPs and 95 CPCs, more than 75% of CPCs were diagnosed in patients younger than 5 years versus 48% of CPPs; and 65% of CPCs and 57% of CPPs occurred in males. In both groups, at least 90% of children underwent surgical resection, and gross total resection (GTR) was achieved in 67% of CPCs and 63.6% of CPPs. Almost 17% of CPCs were treated with radiation versus only 0.9% of CPPs. More than 98% of patients with CPP but only 62% of patients with CPC were alive at last follow-up. [16]
Problem
The choroid plexus is a papillary projection of the ventricular ependyma that is lined by neuroepithelium. Although benign cystic lesions of the choroid plexus are not uncommon, neoplasms are rare. Most choroid plexus neoplasms are benign; however, they can become symptomatic by obstructing CSF flow, eventually leading to generalized increased intracranial pressure or mass effect.
Although patients may be cured by total resection, CPP poses significant surgical risks and challenges related to intraoperative hemostasis. Transcollation devices appear to be an effective and safe addition to the armamentarium of neurosurgical hemostasis options for intracranial tumor resection in which there is high risk of intraoperative hemorrhage. [4]
Epidemiology
Choroid plexus papillomas are rare, accounting for about 1% of all brain tumors, 2-6% of pediatric brain tumors, and 0.5% of adult brain tumors. [11, 12] They most often occur in children, with a predilection for younger ages. [7] CPPs make up 4-6% of intracranial neoplasms in children younger than 2 years and 12-13% of intracranial neoplasms in children younger than 1 year.
CPPs have been associated with von Hippel-Lindau syndrome and Li-Fraumeni syndrome. [17] The frequency of CPPs in children is similar in China (1.5%) [18] and in France (2.3%). [19]
The male-to-female incidence ratio of CPP is 1.2:1.
Etiology
Choroid plexus papillomas arise from the single layer of cuboidal epithelial cells lining the papillae of the choroid plexus. The choroid plexus is associated with the ventricular lining of the body, trigone, and inferior horn of the lateral ventricles; the foramen of Monro; the roof of the third ventricle; and the posterior portion of the roof of the fourth ventricle. Typical locations of the normal choroid plexus correspond to the most common sites for occurrence of CPP.
One study points to the role of a transmembrane receptor protein (Notch3) in the pathogenesis of human choroid plexus tumors. [20] The Notch pathway helps regulate development of the mammalian nervous system, and activation of the Notch pathway has been increasingly recognized in human cancers. Notch3 is expressed in ventricular zone progenitor cells in the fetal brain and, when activated, can function as an oncogene. [16]
Choroid plexus papillomas are associated with the Li-Fraumeni cancer syndrome (an autosomal dominant syndrome characterized by a germline mutation in the TP53 gene) [21] and the Aicardi syndrome (a rare X-linked dominant condition observed in females, characterized by visual impairment, developmental delay, and seizures).
Both somatic and germline abnormalities that involve multiple genetic loci have been associated with the development of choroid plexus tumors. Genomic hybridization data show that choroid plexus papilloma and choroid plexus carcinoma have characteristic chromosomal additions and deletions, which suggests that the genetic basis for these tumors is distinct. [22]
The polyoma viruses SV40, JC, and BK have been implicated in the development of choroid plexus tumors. [23] Choroid plexus tumors have been induced experimentally in transgenic mice by using the polyomavirus common gene product, T antigen. The mechanism is thought to involve binding of T antigen with both pRb and p53 tumor suppressor proteins, as these complexes have been identified in humans with choroid plexus tumors. [24] While research is ongoing to establish the relationship between polyoma viruses and human CNS tumors, further studies have not confirmed this association.
Research has also demonstrated differential expression of several genes in choroid papilloma tumor cells using DNA microarray techniques on cells from 7 choroid plexus papillomas. Among the abnormalities identified was up-regulation of the TWIST-1 transcription factor, which was shown to promote proliferation and in vitro invasion. TWIST-1 is involved in the p53 tumor suppressor pathway as an inhibitor. [25] Genetic analysis of 62 cases of choroid plexus tumors revealed frequent hyperdiploidy in CPPs. Recurrent gains of foci harboring the genes OTX, LAMB1, and TRPM3 were found in all cases in this series. [26]
Relevant Anatomy
Because the choroid plexus is located within the ventricles, the CPP can expand into a space-occupying lesion that may not cause symptoms until either the flow of CSF is blocked or the papilloma becomes large enough to press against the ventricular walls and, subsequently, the brain parenchyma.
These tumors most often occur in the lateral ventricles in children and in the fourth ventricle or the cerebellopontine angle (CPA) in adults. Bilateral CPA choroid plexus papillomas have been reported in the setting of neurofibromatosis type 2. [27] Rarely, CPPs can be found in the third ventricle. Other unusual or rare sites include the sella and primary intraparenchymal sites. [28, 29] Occasionally, CPPs may show extensive calcification or even ossification or may lack their usual radiographic contrast enhancement. [30, 31]
In some instances, the choroid plexus can be found in the CPA, where it has escaped the ventricle via the lateral foramen of Luschka. From this unusual placement of the choroid plexus, or from exophytic growth of the papilloma through the foramen of Luschka, CPPs sometimes manifest in the cerebellopontine angle.
The appearance of CPPs in unusual sites most frequently occurs in the setting of von Hippel-Lindau syndrome.
Grossly, these tumors are tan and lobulated. They fill the ventricles and compress the walls; when they are benign, they do not generally invade brain parenchyma, though they may still rarely seed the CSF.
-
Imaging appearance of a fourth ventricular choroid plexus papilloma (CPP).
-
Histologic appearance of a choroid plexus papilloma (CPP) stained with hematoxylin and eosin.
-
Histologic appearance of a choroid plexus carcinoma stained with hematoxylin and eosin.
Tables

What would you like to print?
- Otolaryngologic Manifestations of Granulomatosis With Polyangiitis
- Pierre Robin Syndrome
- Skill Checkup: Peritonsillar Abscess Drainage
- Biologic Therapies in Refractory Chronic Rhinosinusitis With Nasal Polyps
- Trending Clinical Topic: Tinnitus
- Fast Five Quiz: Do You Know Best Practices for Antibiotic Use in Respiratory Tract Infections?
- Low-Dose CT Vs Chest X-Ray for Lung Cancer Surveillance in HNSCC: Is One Better?
- Does Vitamin D Deficiency Worsen Pediatric Obstructive Sleep Apnea?
- Can ctDNA Accurately Diagnose HPV-Positive Oropharyngeal Squamous Cell Carcinoma?
-
 American Academy of Otolaryngology–Head and Neck Surgery (AAO-HNS) 2024 Annual Meeting
American Academy of Otolaryngology–Head and Neck Surgery (AAO-HNS) 2024 Annual Meeting
-
MSK Pediatrics e-Tumor Boards: Case 2: Multisystem Rosai-Dorfman Disease
-
Managing Common Ear Complaints: An ENT's Advice



 A Lump in the Throat: Thyroid Cancer
A Lump in the Throat: Thyroid Cancer


