Practice Essentials
For patients with thoracic sarcoidosis, when chest radiographic imaging results are correlated with clinical findings, chest radiography may be the only imaging required. Approximately 60-70% of patients with sarcoidosis have characteristic radiologic findings. In 25-30% of patients, radiologic changes are nonspecific or atypical, and in 5-10%, the chest radiograph is normal. Computed tomography (CT) is more sensitive than chest radiography in the detection of mediastinal lymphadenopathy, and high-resolution CT (HRCT) shows subtle parenchymal lung disease with advantage. Left paratracheal, aortopulmonary window, and anterior mediastinal nodes are more readily demonstrated with CT. [1, 2, 3, 4, 5, 6, 7]
Sarcoidosis is a multisystemic granulomatous disease of unknown etiology with variable presentation, prognosis, and progression (see the images below). Sarcoidosis affects an estimated 2 to 160 people per 100,000 worldwide and can involve virtually any organ. Approximately 10-30% of patients with sarcoidosis develop progressive pulmonary disease. Sarcoidosis has a mortality rate of approximately 7% within a 5-year follow-up period. More than 10% of patients with pulmonary sarcoidosis develop progressive disease, and more than 60% of deaths are due to advanced pulmonary sarcoidosis. [8]
Caesar Boeck first coined the term sarcoid in 1899 to describe one of the skin lesions of sarcoidosis because of its histologic resemblance to a sarcoma. In 1905, Boeck described a series of patients with sarcoid who presented with cough and nasal granulomas. These findings suggested the systemic nature of the disease. In 1915, Kusnitski and Bittorf described chest radiographic abnormalities in a patient with sarcoidosis.
(See the images below.)
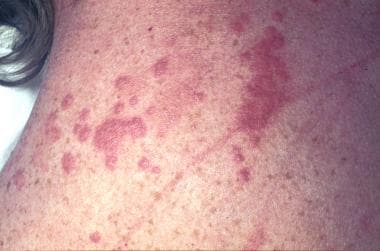
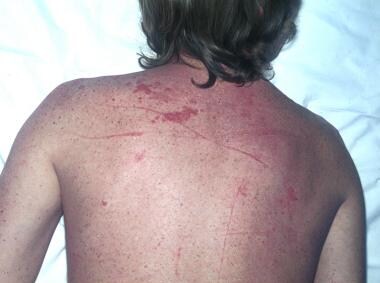 Sarcoidosis, thoracic. Crops of numerous papules on the back of 33-year-old woman. Some papules have become confluent. Note that the skin in between the papules is of normal color. These lesions are usually associated with a good prognosis in sarcoidosis. Note also the scar sarcoid. Scars become infiltrated and purple, resembling keloids. These occur in active disease.
Sarcoidosis, thoracic. Crops of numerous papules on the back of 33-year-old woman. Some papules have become confluent. Note that the skin in between the papules is of normal color. These lesions are usually associated with a good prognosis in sarcoidosis. Note also the scar sarcoid. Scars become infiltrated and purple, resembling keloids. These occur in active disease.
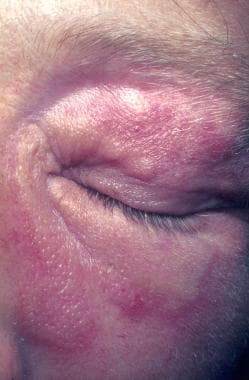 Sarcoidosis, thoracic. Lupus pernio affects poorly perfused areas, such as the nose, ear lobes, and fingers. These areas become swollen and indurated, with deep purplish red. The lesions are often associated bone cysts due to sarcoid granulomas, and the nasal bones may be eroded. In this 38-year-old woman, the lesions affect the periorbital areas and the nose. The fingers were also affected (not shown).
Sarcoidosis, thoracic. Lupus pernio affects poorly perfused areas, such as the nose, ear lobes, and fingers. These areas become swollen and indurated, with deep purplish red. The lesions are often associated bone cysts due to sarcoid granulomas, and the nasal bones may be eroded. In this 38-year-old woman, the lesions affect the periorbital areas and the nose. The fingers were also affected (not shown).
Sarcoidosis almost always affects the respiratory system. Most patients present with the classic combination of bilateral hilar lymphadenopathy, parenchymal disease of the lung, and eye or skin lesions; however, virtually any organ in the body may be involved. At diagnosis, about 50% of patients are asymptomatic, 25% complain of cough or dyspnea, and 25% have or develop eye symptoms or skin lesions (eg, erythema nodosum, lupus pernio, plaques, scars). When present, constitutional symptoms include weight loss, fatigue, weakness, and malaise. Symptoms of pulmonary involvement such as dry cough and shortness of breath develop in 20-30% of patients. [9, 10] In general, sarcoidosis is a self-limiting subclinical process in 60-70% of cases. About 20-30% of patients are left with a variable degree of permanent lung damage. In 10-15%, sarcoidosis can become chronic. The incidence of ocular involvement is about 20-30%.
Bilateral hilar lymphadenopathy is the most common radiographic finding. Other radiographic findings include interstitial lung disease, occasional calcification of affected lymph nodes, and, rarely, pleural effusions and thickening.
Because the disease so often involves thoracic structures, chest radiography plays a crucial role in the diagnosis, staging, and follow-up of sarcoidosis. The diagnosis is established when clinical and radiographic findings are supported by histologic evidence of widespread noncaseating epithelioid cell granulomas in more than 1 organ or a positive Kveim-Stiltzbach skin test result.
(See the images below.)
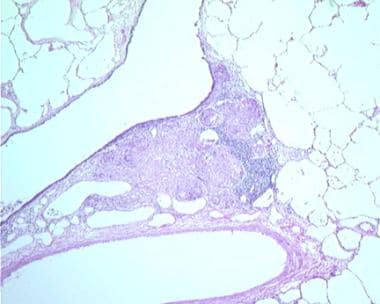 Sarcoidosis, thoracic. Series of histologic slides (see the next 2 images) from a patient with sarcoidosis show characteristic noncaseating granulomas with many giant cells. Courtesy of Sat Sharma, MD, FRCPC, FCCP, DABSM.
Sarcoidosis, thoracic. Series of histologic slides (see the next 2 images) from a patient with sarcoidosis show characteristic noncaseating granulomas with many giant cells. Courtesy of Sat Sharma, MD, FRCPC, FCCP, DABSM.
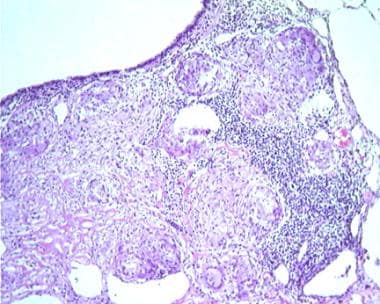 Sarcoidosis, thoracic. Series of histologic slides (see the previous image and the next image) from a patient with sarcoidosis show characteristic noncaseating granulomas with many giant cells. Courtesy of Sat Sharma, MD, FRCPC, FCCP, DABSM.
Sarcoidosis, thoracic. Series of histologic slides (see the previous image and the next image) from a patient with sarcoidosis show characteristic noncaseating granulomas with many giant cells. Courtesy of Sat Sharma, MD, FRCPC, FCCP, DABSM.
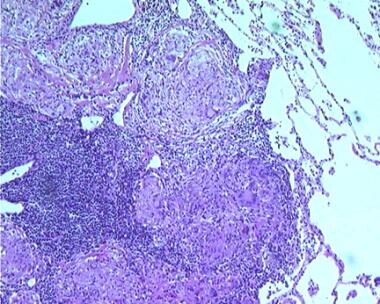 Sarcoidosis, thoracic. Series of histologic slides (see the previous 2 images) from a patient with sarcoidosis show characteristic noncaseating granulomas with many giant cells. Courtesy of Sat Sharma, MD, FRCPC, FCCP, DABSM.
Sarcoidosis, thoracic. Series of histologic slides (see the previous 2 images) from a patient with sarcoidosis show characteristic noncaseating granulomas with many giant cells. Courtesy of Sat Sharma, MD, FRCPC, FCCP, DABSM.
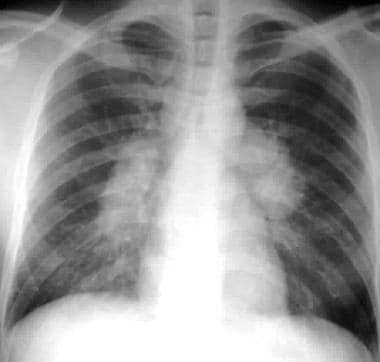 Sarcoidosis, thoracic. Stage I disease. Standard posteroanterior chest radiograph in a 28-year-old man shows extensive bilateral hilar and mediastinal lymph node enlargement not associated with a pulmonary abnormality.
Sarcoidosis, thoracic. Stage I disease. Standard posteroanterior chest radiograph in a 28-year-old man shows extensive bilateral hilar and mediastinal lymph node enlargement not associated with a pulmonary abnormality.
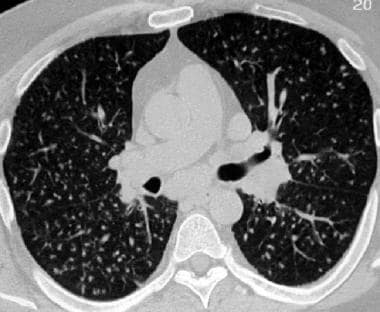 Sarcoidosis, thoracic. High-resolution CT scan in a young patient shows uniformly small, bilateral nodules in a miliary pattern. The patient also had mediastinal and hilar adenopathy. This is stage II disease.
Sarcoidosis, thoracic. High-resolution CT scan in a young patient shows uniformly small, bilateral nodules in a miliary pattern. The patient also had mediastinal and hilar adenopathy. This is stage II disease.
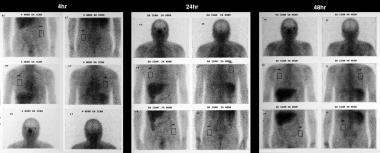 Sarcoidosis, thoracic. Gallium-67 scans in a patient who had a normal chest radiograph. Study shows increased uptake in the lung fields, higher than the background activity. The appearances are compatible with pneumonitis secondary to sarcoidosis.
Sarcoidosis, thoracic. Gallium-67 scans in a patient who had a normal chest radiograph. Study shows increased uptake in the lung fields, higher than the background activity. The appearances are compatible with pneumonitis secondary to sarcoidosis.
Staging
Clinical staging of sarcoidosis is based on the pattern of chest radiographic findings:
-
Stage 0 is a normal chest radiograph
-
Stage I is lymphadenopathy only
-
Stage II is lymphadenopathy and lung parenchymal disease
-
Stage III is parenchymal lung disease only
-
Stage IV is pulmonary fibrosis
At presentation, approximately 5-10% of patients have stage 0 disease; 50%, stage I; 25-30%, stage II; and 15%, stage III. Although most cases of sarcoidosis either regress or remain stable, 10-15% progress to pulmonary fibrosis. Generally, pulmonary function worsens with an increasing stage of disease, but radiologic staging does not correlate well with the severity of pulmonary function abnormalities. Often, radiographic abnormalities appear worse than the degree of functional impairment that is actually present.
Imaging modalities
When chest radiographic results are correlated with clinical findings, chest radiography may be the only imaging required. [1, 11] Routine chest CT scanning is not usually indicated and adds little to patient care.
Approximately 60-70% of patients with sarcoidosis have characteristic radiologic findings. In 25-30% of patients, radiologic changes are nonspecific or atypical, and in 5-10%, the chest radiograph is normal. Thus, approximately 30-40% of patients can benefit from a CT scan. CT is more sensitive than radiography in detection of lymphadenopathy and subtle parenchymal disease. High-resolution CT (HRCT) is useful in differentiating active disease from fibrosis. HRCT results are well correlated with the yield of biopsy.
Diagnosis of sarcoidosis relies on a consistent clinical picture, histologic demonstration of noncaseating granulomas, and exclusion of other diseases with a similar histologic or clinical picture. Nevertheless, chest imaging has an important role in the diagnosis (allowing clinicians to avoid biopsy in some situations) and prognosis of sarcoidosis. [12]
Accumulation of gallium-67 (67Ga) is a sensitive but nonspecific indicator of active inflammation in patients with sarcoidosis. Gallium-67 avidity alone cannot be used to establish a diagnosis of sarcoidosis, and this finding has a limited correlation with the patient's clinical status. However, 67Ga scintigraphy is useful in identifying extrathoracic sites of involvement, in detecting active alveolitis, and in assessing response to treatment. Gallium-67 scanning can be helpful when the clinical picture remains confusing despite the presence of noncaseating granulomas in biopsy specimens, and it may be useful in differentiating chronic hypersensitivity pneumonitis from sarcoidosis. [13, 14, 15, 16, 17]
Mycetomas may occur in more than 50% of patients with stage IV sarcoidosis and apical bullous disease. Although mycetomas may be clinically silent, hemoptysis is common. Life-threatening hemoptysis may be managed with angiographic localization of bleeding and concurrent bronchial artery embolization in a minority of patients. Bronchial necrosis is more frequently encountered when absolute alcohol is used for embolization. The diagnosis is most confidently established when clinicoradiologic findings are supported by histologic evidence of widespread noncaseating granulomas.
Limitations of techniques
The limitation of all techniques is the nonspecificity of most methods of diagnostic imaging. A chest radiograph can be normal in biopsy-proven sarcoidosis. A false-positive diagnosis may occur with a variety of granulomatous diseases. Problems may also arise with the biopsy material.
CT is expensive and has a radiation burden. Gallium-67 scanning is limited by nonspecificity, and scans may show normal uptake in established disease. Gallium-67 scanning is also time-consuming and expensive.
Okada et al concluded that a chest radiograph and serum angiotensin-converting enzyme (ACE) levels sufficiently reflect disease activity and that routine evaluation with 67Ga scanning and bronchoalveolar lavage is not necessarily indicated in the long-term management of patients with pulmonary sarcoidosis. [18]
Radiography
Radiographic staging
Characteristic radiographic appearances are reported in approximately 60-70% of patients with sarcoidosis. [19]
Radiographic changes in thoracic sarcoidosis are usefully classified into 5 groups or stages, as follows:
-
Stage 0 - No demonstrable abnormality
-
Stage 1 - Hilar and mediastinal lymph node enlargement not associated with pulmonary abnormality (see the image below)
 Sarcoidosis, thoracic. Stage I disease. Standard posteroanterior chest radiograph in a 28-year-old man shows extensive bilateral hilar and mediastinal lymph node enlargement not associated with a pulmonary abnormality.
Sarcoidosis, thoracic. Stage I disease. Standard posteroanterior chest radiograph in a 28-year-old man shows extensive bilateral hilar and mediastinal lymph node enlargement not associated with a pulmonary abnormality.
-
Stage 2 - Hilar and mediastinal lymph node enlargement associated with pulmonary abnormality (see the images below)
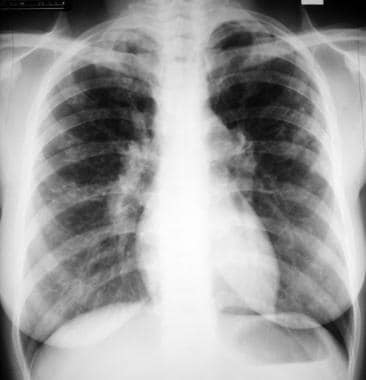 Sarcoidosis, thoracic. Stage II disease. Chest radiograph in a 36-year-old woman shows mediastinal lymph node enlargement and bilateral pulmonary opacities.
Sarcoidosis, thoracic. Stage II disease. Chest radiograph in a 36-year-old woman shows mediastinal lymph node enlargement and bilateral pulmonary opacities.
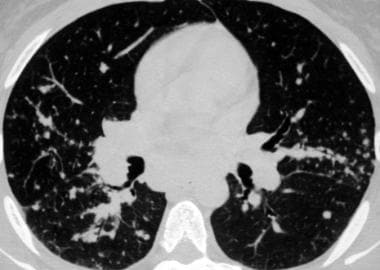 Sarcoidosis, thoracic. Pulmonary window CT image in the same patient as in the previous image shows small nodules mostly along the bronchovascular bundles, giving the bronchi and vessels a beaded appearance. This distribution along the bronchovascular bundles accounts for the fact that transbronchial biopsy is usually successful for obtaining tissue for diagnosis.
Sarcoidosis, thoracic. Pulmonary window CT image in the same patient as in the previous image shows small nodules mostly along the bronchovascular bundles, giving the bronchi and vessels a beaded appearance. This distribution along the bronchovascular bundles accounts for the fact that transbronchial biopsy is usually successful for obtaining tissue for diagnosis.
-
Stage 3 - Diffuse pulmonary disease not associated with nodal enlargement (see the images below)
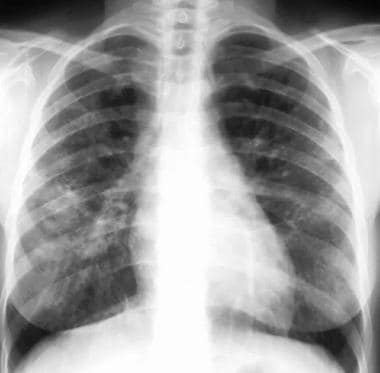 Sarcoidosis, thoracic. Posteroanterior (PA) chest radiograph in a 38-year-old man shows stage III disease associated with a tumefactive type of lung parenchymal involvement: opacities in the right and left midlung zones that mimic neoplasm.
Sarcoidosis, thoracic. Posteroanterior (PA) chest radiograph in a 38-year-old man shows stage III disease associated with a tumefactive type of lung parenchymal involvement: opacities in the right and left midlung zones that mimic neoplasm.
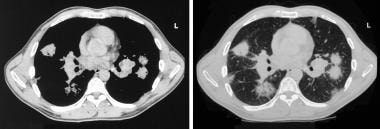 Sarcoidosis, thoracic. CT axial images through the thorax in the same patient as in the previous image. Left, Mediastinal window setting shows bilateral hilar adenopathy. Right, Pulmonary window image shows round pulmonary opacities.
Sarcoidosis, thoracic. CT axial images through the thorax in the same patient as in the previous image. Left, Mediastinal window setting shows bilateral hilar adenopathy. Right, Pulmonary window image shows round pulmonary opacities.
-
Stage 4 - Pulmonary fibrosis




Hilar and mediastinal lymphadenopathy
Hilar and/or mediastinal lymphadenopathy is found in the majority of patients with sarcoidosis; this is the most common finding. The most frequent presentation is bilateral hilar and right paratracheal lymphadenopathy. Left paratracheal and aortopulmonary window lymph nodes are also frequently involved, but they may be difficult to detect on a standard posteroanterior (PA) chest radiographs.
Atypical lymphadenopathy may rarely affect the paratracheal, subcarinal, aortopulmonary window, and retroazygous nodes unaccompanied by hilar lymphadenopathy. Radiographic evidence of anterior mediastinal adenopathy is seen in less than 10% of cases. The posterior mediastinum is least commonly involved. Findings of isolated lymphadenopathy in the anterior or posterior mediastinal compartments should raise the possibility of diagnoses other than sarcoidosis. Isolated unilateral hilar lymphadenopathy is an unusual manifestation of sarcoidosis, occurring in only 1-3% of patients.
Patients older than 50 years may present with atypical mediastinal lymphadenopathy. Mediastinal lymphadenopathy with no associated hilar lymphadenopathy or unilateral hilar lymphadenopathy occurs more frequently in such older patients. Patients older than 50 years also have a higher prevalence of solitary and multiple masslike opacities in the lung at their first presentation. In such older patients, the prevalence of atelectasis may be higher.
Dystrophic calcification
Dystrophic calcification of involved lymph nodes is related to duration of disease, occurring in 3% of cases after 5 years and in 20% after 10 years. Calcification can be amorphous, punctate, popcornlike, or eggshell. [20]
Parenchymal changes
Parenchymal changes from interstitial lung involvement may mimic airspace disease. On plain radiographs, parenchymal disease may show a variety of radiographic patterns, including fine nodular; reticulonodular; acinar (poorly marginated, small to large nodules or coalescent opacities); and, rarely, focal (solitary nodule or mass). In sarcoidosis, acinar opacities or interstitial granulomas may coalesce to give the appearance of the alveolar form of sarcoidosis, and an air bronchogram may be exhibited.
Cavitation of sarcoid parenchymal lesions rarely occurs; it is found in less than 1% of patients. When cavitation occurs, tuberculous and fungal infections need to be ruled out. Necrotizing sarcoid granulomatosis is a variant of sarcoidosis that is predominantly an angiitis rather than an alveolitis; it is more likely to give rise to cavitating pulmonary nodules, particularly in a peripheral, pleural-based location.
Mycetomas
Mycetomas develop in more than 50% of patients with stage IV sarcoidosis and apical bullous disease. The earliest manifestations of mycetoma formation are adjacent pleural thickening or apparent thickening of the wall of a bulla. Later, an intracavitary, gravity-dependent mass (Mounod sign) may be seen. Although mycetomas may be clinically silent, hemoptysis is common. Life-threatening hemoptysis that requires surgical intervention or angiographic localization of bleeding with concurrent bronchial artery embolization occurs in a minority of patients.
Lofgren syndrome
Lofgren syndrome is a clinically distinct phenotype of sarcoidosis and is a multisystem granulomatous disorder that commonly involves the lungs and, secondly, the skin. The cutaneous manifestations are seen in up to 33% of patients and may be the first sign of the disease. Lofgren syndrome presents acutely and typically presents in younger patients with acute onset of erythema nodosum, bilateral hilar lymphadenopathy, fever, and migratory polyarthritis but without granulomatous skin involvement. The classic triad of fever, erythema nodosum, and bilateral hilar adenopathy, however, is not always present and has also been associated with migratory polyarthritis, especially involving the ankles in men. It is important for clinicians to recognize Lofgren syndrome and to differentiate it from the classic presentation of sarcoidosis, because this syndrome can have different diagnostic and therapeutic consequences, including permanent scarring of the lungs if left untreated. [21, 22]
Other findings
Involvement of the pleura by the granulomatous process may result in small to moderate effusions. These effusions usually resolve in 2-3 months, as shown on radiographs. Pleural thickening may result and usually involves the lower chest.
In stage IV sarcoidosis, when fibrosis supervenes, hilar retraction, decreased lung volume, and honeycomb lung may be present. Bullous disease airtrapping and diaphragmatic tenting may also be seen.
Pulmonary arterial hypertension and right heart failure may develop as a result of extensive interstitial fibrosis. Radiographic findings include a prominent main pulmonary artery, enlarged right and left pulmonary arteries, right ventricular enlargement, and attenuation of peripheral vessels.
Any part of the airway can be affected, from the epiglottis to the bronchioles. Tracheal stenosis is rare. Mediastinal lymphadenopathy may extrinsically compress bronchi, or bronchial obstruction may result from endobronchial granulomas. Bronchiectasis or frank occlusion and/or stenosis may develop as a result of scarring and fibrosis.
Cardiac involvement is usually difficult to detect on plain radiography. Radiologic signs of cardiac sarcoidosis are nonspecific and include enlarged cardiopericardial silhouette, signs of pulmonary venous hypertension, arterial pulmonary hypertension, and heart failure.
Health care providers who care for older adults should be aware of the increasing prevalence of late-onset sarcoidosis and should consider the diagnosis in those who present with otherwise unexplained systemic symptoms, thoracic abnormalities on imaging, and/or evidence of other organ involvement. Providing earlier diagnosis and therapeutic intervention to halt the development of pulmonary fibrosis and pulmonary hypertension and monitoring for treatment-related adverse effects will confer a mortality benefit. [23]
Degree of confidence
Chest radiography is a noninvasive test that is universally available. When correlated with clinical findings, it may be the only imaging required. In most patients, sarcoidosis has a characteristic appearance on plain chest radiographs. In approximately one quarter of patients, radiologic changes are nonspecific or atypical, and the chest radiograph is normal in a minority. However, the course of parenchymal disease is unpredictable, and no radiographic criteria exist to distinguish reversible from irreversible parenchymal changes until irreversible fibrosis has been long standing.
Miller and Putman compared chest radiographs with gallium-67 scans in 85 studies in 51 patients with biopsy-proven sarcoidosis [24] and found that chest radiography compared favorably with other staging methods in detecting the alveolitis phase of sarcoidosis and that the examination was reproducible, noninvasive, and cost-efficient. [19]
False positives/negatives
The chest radiograph may be normal in 5-10% of patients. In 25-30%, radiologic changes are nonspecific or atypical. In some patients, atypical lymphadenopathy lymphomas can mimic fungal infections, tuberculosis, and cancer.
Left paratracheal and aortopulmonary window lymph nodes are less frequently involved, but they may be difficult to detect on a standard posteroanterior chest radiograph. End-stage lung parenchymal disease may be indistinguishable from lung disease of many other causes. A miliary pattern on a chest radiograph can be indistinguishable from patterns seen with tuberculosis, fungal infections, histiocytosis, and miliary metastases. Sarcoidosis rarely causes pleural effusions; when they do occur, they can be indistinguishable from pleural effusions of other causes.
Computed Tomography
High-resolution CT
Computed tomography (CT) is more sensitive than chest radiography in detection of mediastinal lymphadenopathy, and high-resolution CT (HRCT) shows subtle parenchymal lung disease with advantage. Left paratracheal, aortopulmonary window, and anterior mediastinal nodes are more readily demonstrated with CT. With 1- to 1.5-mm sections and a high-spatial-frequency reconstruction algorithm, HRCT produces excellent images of anatomic regions affected by the granulomatous process. Early acinar patterns, parenchymal nodules, and nodular consolidation are more clearly defined on these scans than on chest radiographs. [1, 2, 3, 4, 5, 6, 7, 25]
In sarcoidosis, HRCT findings include areas of ground-glass attenuation; subpleural nodules; perivascular nodules, which appear as beading and irregular thickening of bronchovascular bundles; and thickening of interlobular septa. The nodules, which correspond to coalescent interstitial granulomas, have irregular margins. The foci of ground-glass attenuation may represent areas of active alveolitis or diffuse microscopic interstitial granulomas that cannot be demonstrated on HRCT scans (see the images below). [26, 27, 28, 29]
In a study by Zhang et al, researchers found that 50.2% of patients with confirmed pulmonary sarcoidosis showed discordance in sarcoidosis stage between chest x-ray (CXR) and HRCT. According to the authors, the primary reason for the inconsistency was failure of radiographs to detect mediastinal lymph node enlargement in the shadow of the heart, along with failure to detect small nodules because of limited x-ray resolution. Pleural involvement was detected by HRCT in 25.6% of patients but in only 7.5% by radiography. [30]
Sarcoidosis, thoracic. High-resolution CT scan in a young patient shows uniformly small, bilateral nodules in a miliary pattern. The patient also had mediastinal and hilar adenopathy. This is stage II disease.
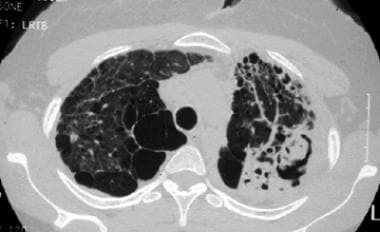 Sarcoidosis, thoracic. High-resolution CT scan in a young woman with a several-year history of sarcoidosis shows lung destruction with air-spaces, focal consolidation, and a fungus ball in a cavity in the left lower lung. This is stage IV disease.
Sarcoidosis, thoracic. High-resolution CT scan in a young woman with a several-year history of sarcoidosis shows lung destruction with air-spaces, focal consolidation, and a fungus ball in a cavity in the left lower lung. This is stage IV disease.
Stage IV sarcoidosis is often associated with upper lung fibrosis, which is depicted well with HRCT. Findings include honeycombing, bullae and cyst formation, and bronchiectasis. HRCT findings of fibrosis include lung distortion with posterior displacement of main and upper lobe bronchi, traction bronchiectasis, abnormal central conglomeration of hilar and perihilar structures, and upper lobe conglomerate masses (see the images below).
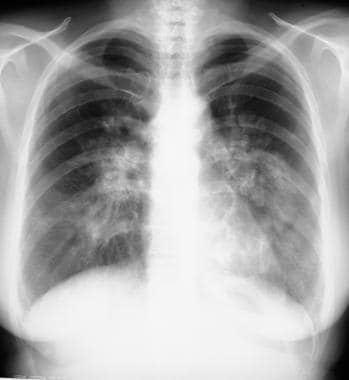 Sarcoidosis, thoracic. Posteroanterior chest radiograph shows enlarged calcified hilar lymph nodes with calcifications.
Sarcoidosis, thoracic. Posteroanterior chest radiograph shows enlarged calcified hilar lymph nodes with calcifications.
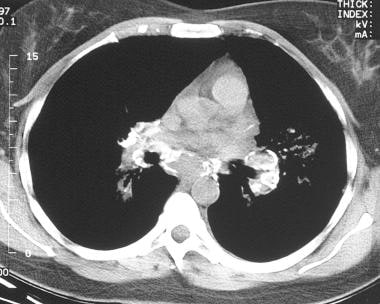 Sarcoidosis, thoracic. Axial CT scan through the mediastinum in the same patient as in the previous image shows extensive calcifications in the mediastinal and hilar lymph nodes.
Sarcoidosis, thoracic. Axial CT scan through the mediastinum in the same patient as in the previous image shows extensive calcifications in the mediastinal and hilar lymph nodes.
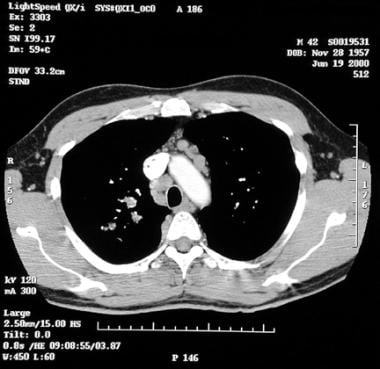 Sarcoidosis, thoracic. Contrast-enhanced axial CT image at the level of the aorta shows enlarged pretracheal and prevascular lymph nodes.
Sarcoidosis, thoracic. Contrast-enhanced axial CT image at the level of the aorta shows enlarged pretracheal and prevascular lymph nodes.
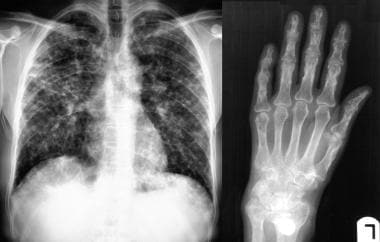 Sarcoidosis, thoracic. Posteroanterior chest radiograph shows extensive lung parenchymal involvement. At this stage, the patient had moderate-effort dyspnea. Radiograph of the right hand in the same patient shows extensive sarcoid osseous lesions in the phalanges and metacarpals.
Sarcoidosis, thoracic. Posteroanterior chest radiograph shows extensive lung parenchymal involvement. At this stage, the patient had moderate-effort dyspnea. Radiograph of the right hand in the same patient shows extensive sarcoid osseous lesions in the phalanges and metacarpals.
Degree of confidence
CT can sometimes demonstrate focal parenchymal involvement in patients with stage 0 or 1 radiographic findings. However, HRCT results may be normal in the presence of microscopic disease. In patients with sarcoidosis, CT findings include relatively symmetric and diffuse involvement of mediastinal and hilar lymph nodes, as well as thickened bronchovascular bundles.
To a lesser extent, subpleural small nodules may be seen, along with interlobular septal thickening. Centrilobular attenuating areas can be detected as thickened peripheral bronchovascular bundles. On CT scans, ground-glass areas usually represent an accumulation of many granulomas in the interstitium, but they may not indicate an alveolitis.
False positives/negatives
Hilar and/or mediastinal lymphadenopathy is not specific for sarcoidosis; similar findings may occur in lymphoma, leukemia, metastases, and fungal and viral infections.
With HRCT, the classification of diffuse lung parenchymal disease is based on 4 distributions: centrilobular, perilobular, panlobular, and nonlobular.
The centrilobular classification is seen in airway diseases such as Mycoplasma pneumonia, tuberculosis, bronchiolitis, and interstitial lung diseases (eg, sarcoidosis, lymphangitic carcinomatosis, chronic interstitial pneumonias).
Perilobular patterns are seen in sarcoidosis, lymphangitic carcinomatosis, non-Hodgkin lymphoma, and chronic interstitial pneumonias.
A panlobular distribution is seen in advanced stages of interstitial lung disease, such as lymphangitic carcinomatosis and sarcoidosis.
A nonlobular pattern is typically seen in Pneumocystis jirovecii pneumonia but may occur in other interstitial lung diseases such as sarcoidosis.
Magnetic Resonance Imaging
Mendelson and associates examined 15 patients with sarcoidosis by using magnetic resonance imaging (MRI) and found that the T2 signal intensity of mediastinal lymphadenopathy varied, with no characteristic pattern noted. Three of 4 patients with bright lymph nodes on T2-weighted images had stage I disease, but low-intensity lymph nodes were also seen. In their study, subcarinal nodes were best depicted on coronal images. [31]
MRI is also useful in characterizing osseous involvement with sarcoid, particularly in the spine. [32, 33]
As yet, MRI has not supplanted CT in the evaluation of thoracic sarcoidosis. Technical difficulties still exist with MRI, and cardiorespiratory movement remains a problem in thoracic imaging. In patients with chronic infiltrative lung disease, MRI appears to be equal to CT in the demonstration of areas of airspace opacification, but it is inferior to CT in assessment of fine lung-parenchymal anatomy or fibrosis. [34]
MRI has a potential role in the evaluation of pericardial and/or myocardial involvement. MRI is not useful in distinguishing the lymphadenopathy of sarcoidosis from that of other entities, but it is useful for defining the anatomic extent of the disease and for differentiating enlargement of the pulmonary artery from lymphadenopathy.
MRI cannot be used to distinguish mediastinal sarcoid lymphadenopathy from mediastinal lymph node enlargement that results from other causes.
Positron Emission Tomography
Positron emission tomography (PET) has emerged as a method to determine the location and extent of disease activity in sarcoidosis. Most clinicians do not routinely utilize PET in the management of sarcoidosis, but it is important to understand the impact that abnormalities detected by PET can have on diagnosis, prognosis, and treatment. [35]
PET is most often utilized for the diagnosis of cardiac sarcoidosis, for which it may provide information about prognosis and adverse events. PET combined with cardiac magnetic resonance (CMR) may enable clinicians to increase the diagnostic yield of imaging. Furthermore, PET abnormalities can be utilized in reduction or augmentation of therapy based on an individual's response to treatment. [35]
PET performed with 18F-fluorodeoxyglucose (FDG) can play a role in lesion detection, identification of accessible biopsy sites, quantification of disease activity, prognostication of outcome, and response assessment in patients with sarcoidosis. [36, 37] Prospective trials are needed to define the role of PET and to standardize its performance and interpretation. [35]
Nuclear Imaging
Gallium-67–avid disease has been reported in more than 90% of cases of pulmonary involvement. A lambda pattern of uptake in the parahilar, infrahilar bronchopulmonary, and mediastinal lymph nodes has been described in 72% of patients with intrathoracic sarcoidosis. Symmetric uptake in the parotid and lacrimal glands occurs in 79% of patients (see the images below). [38, 13, 14, 15, 16, 17]
Sarcoidosis, thoracic. Gallium-67 scans in a patient who had a normal chest radiograph. Study shows increased uptake in the lung fields, higher than the background activity. The appearances are compatible with pneumonitis secondary to sarcoidosis.
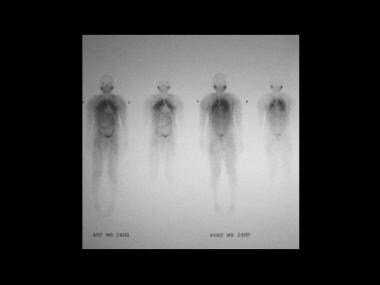 Sarcoidosis, thoracic. Gallium-67 scans in a patient with biopsy-proved lung sarcoidosis. Note intense activity in the lung fields suggestive of pneumonitis secondary to sarcoidosis.
Sarcoidosis, thoracic. Gallium-67 scans in a patient with biopsy-proved lung sarcoidosis. Note intense activity in the lung fields suggestive of pneumonitis secondary to sarcoidosis.
One or both of the aforementioned patterns of uptake occur in approximately 90% of patients. This pattern of activity does not usually occur in patients with lymphoma. The only exception is seen in patients who have undergone head and neck radiation therapy and have subsequently developed radiation sialoadenitis. Scans in these patients may show symmetric 67Ga uptake that produces the panda sign. [39, 40, 41]
In a study by Sharma et al, Ga-DOTA-NOC PET/CT was found to be of help in assessing disease activity and treatment response in patients with sarcoidosis with thoracic involvement. The maximum standardized uptake value (SUVmax) at the pathologic site and in the descending thoracic aorta (SUVmed) were assessed. A SUVmax/SUVmed ratio (disease activity score) of more than 1 was considered a marker of active disease. In 39 patients (27 symptomatic, 12 asymptomatic), increased disease activity was present in 25 (92%) of the 27 symptomatic patients and 2 (16%) of the 12 asymptomatic patients. Sensitivity and specificity of the test were 92.5% and 83.3%, respectively. In addition, of 9 patients who became asymptomatic after treatment, 7 showed a significant decrease in mean disease activity. [14]
Degree of confidence
Gallium-67 scanning is more sensitive than chest radiography in determining the degree and variation of pulmonary sarcoidosis activity, in evaluating response to therapy, and in foreseeing relapses. Gallium-67–avid disease has been reported in more than 90% of cases of pulmonary involvement, although 67Ga avidity is nonspecific. However, 67Ga scans may be useful as a baseline study at the time of diagnosis. If results of 67Ga scintigraphy are initially positive, negative findings from a subsequent 67Ga scan obtained during the course of treatment suggest that alveolitis has resolved.
Accumulation of 67Ga is a sensitive but nonspecific indicator of active inflammation in patients with sarcoidosis. Gallium-67 avidity cannot be used alone to establish a diagnosis of sarcoidosis, and it has limited correlation with clinical status. However, 67Ga scintigraphy is useful in identifying extrathoracic sites of involvement, detecting active alveolitis, and assessing treatment response.
Gallium-67 scans have low sensitivity and specificity as a diagnostic test. Gallium-67 scanning is useful for patients in whom the clinical picture remains confusing despite the presence of noncaseating granulomas in biopsy specimens, and scanning may useful in differentiating chronic hypersensitivity pneumonitis from sarcoidosis. Gallium-67 avidity alone cannot be used to establish a diagnosis of sarcoidosis.
However, in several studies, FDG PET and PET/CT have been demonstrated to be more sensitive than 67Ga. For example, Braun et al demonstrated sensitivity of 79% for FDG PET/CT in biopsy-proven sites compared with 58% for 67Ga. The sensitivity of PET/CT was 86% when skin sites were excluded. [42]
Rubini et al found that for restaging and follow-up of sarcoidosis, FDG PET/CT had a sensitivity, specificity, and accuracy of 100%, 50%, and 87.5%, respectively, as compared to 92%, 81%, and 50%, respectively, for CT. [43]
False positives/negatives
Gallium-67 is normally localized in the liver, spleen, bone marrow, bone, and growth plates. A lesser degree of normal accumulation occurs in the salivary and lacrimal glands and in breast tissue. Gut activity is related to small-bowel excretion and, in part, biliary excretion. Hodgkin and non-Hodgkin lymphomas, lung cancers, melanomas, infections, and inflammations are 67Ga avid, and this can potentially cause false-positive results for sarcoidosis.
False-negative 67Ga scans may occur in up to 10% of patients. In patients with lymphoma who have undergone head and neck radiation therapy, uptake of 67Ga in the neck may be similar to that in patients with sarcoidosis.
-
Sarcoidosis, thoracic. Crops of numerous papules on the back of 33-year-old woman. Some papules have become confluent. Note that the skin in between the papules is of normal color. These lesions are usually associated with a good prognosis in sarcoidosis. Note also the scar sarcoid. Scars become infiltrated and purple, resembling keloids. These occur in active disease.
-
Sarcoidosis, thoracic. Close-up view of the previous image.
-
Sarcoidosis, thoracic. Lupus pernio affects poorly perfused areas, such as the nose, ear lobes, and fingers. These areas become swollen and indurated, with deep purplish red. The lesions are often associated bone cysts due to sarcoid granulomas, and the nasal bones may be eroded. In this 38-year-old woman, the lesions affect the periorbital areas and the nose. The fingers were also affected (not shown).
-
Sarcoidosis, thoracic. Series of histologic slides (see the next 2 images) from a patient with sarcoidosis show characteristic noncaseating granulomas with many giant cells. Courtesy of Sat Sharma, MD, FRCPC, FCCP, DABSM.
-
Sarcoidosis, thoracic. Series of histologic slides (see the previous image and the next image) from a patient with sarcoidosis show characteristic noncaseating granulomas with many giant cells. Courtesy of Sat Sharma, MD, FRCPC, FCCP, DABSM.
-
Sarcoidosis, thoracic. Series of histologic slides (see the previous 2 images) from a patient with sarcoidosis show characteristic noncaseating granulomas with many giant cells. Courtesy of Sat Sharma, MD, FRCPC, FCCP, DABSM.
-
Sarcoidosis, thoracic. Stage I disease. Standard posteroanterior chest radiograph in a 28-year-old man shows extensive bilateral hilar and mediastinal lymph node enlargement not associated with a pulmonary abnormality.
-
Sarcoidosis, thoracic. Stage II disease. Chest radiograph in a 36-year-old woman shows mediastinal lymph node enlargement and bilateral pulmonary opacities.
-
Sarcoidosis, thoracic. Pulmonary window CT image in the same patient as in the previous image shows small nodules mostly along the bronchovascular bundles, giving the bronchi and vessels a beaded appearance. This distribution along the bronchovascular bundles accounts for the fact that transbronchial biopsy is usually successful for obtaining tissue for diagnosis.
-
Sarcoidosis, thoracic. Posteroanterior (PA) chest radiograph in a 38-year-old man shows stage III disease associated with a tumefactive type of lung parenchymal involvement: opacities in the right and left midlung zones that mimic neoplasm.
-
Sarcoidosis, thoracic. CT axial images through the thorax in the same patient as in the previous image. Left, Mediastinal window setting shows bilateral hilar adenopathy. Right, Pulmonary window image shows round pulmonary opacities.
-
Sarcoidosis, thoracic. High-resolution CT scan in a young patient shows uniformly small, bilateral nodules in a miliary pattern. The patient also had mediastinal and hilar adenopathy. This is stage II disease.
-
Sarcoidosis, thoracic. High-resolution CT scan in a young woman with a several-year history of sarcoidosis shows lung destruction with air-spaces, focal consolidation, and a fungus ball in a cavity in the left lower lung. This is stage IV disease.
-
Sarcoidosis, thoracic. Posteroanterior chest radiograph shows enlarged calcified hilar lymph nodes with calcifications.
-
Sarcoidosis, thoracic. Axial CT scan through the mediastinum in the same patient as in the previous image shows extensive calcifications in the mediastinal and hilar lymph nodes.
-
Sarcoidosis, thoracic. Contrast-enhanced axial CT image at the level of the aorta shows enlarged pretracheal and prevascular lymph nodes.
-
Sarcoidosis, thoracic. Posteroanterior chest radiograph shows extensive lung parenchymal involvement. At this stage, the patient had moderate-effort dyspnea. Radiograph of the right hand in the same patient shows extensive sarcoid osseous lesions in the phalanges and metacarpals.
-
Sarcoidosis, thoracic. Gallium-67 scans in a patient who had a normal chest radiograph. Study shows increased uptake in the lung fields, higher than the background activity. The appearances are compatible with pneumonitis secondary to sarcoidosis.
-
Sarcoidosis, thoracic. Gallium-67 scans in a patient with biopsy-proved lung sarcoidosis. Note intense activity in the lung fields suggestive of pneumonitis secondary to sarcoidosis.
